Chapter 119 Primary Defects of Cellular Immunity
In general, patients with defects in T-cell function have infections or other clinical problems that are more severe than in patients with antibody deficiency disorders (see Table 116-4). The defective gene products for some primary T-cell diseases are identified (Table 119-1). These individuals rarely survive beyond infancy or childhood. Transplantation of thymic tissue, or of major histocompatibility complex (MHC)-compatible sibling or haploidentical (half-matched) parental hematopoietic stem cell, is the treatment of choice for patients with primary T-cell defects (Chapter 129).
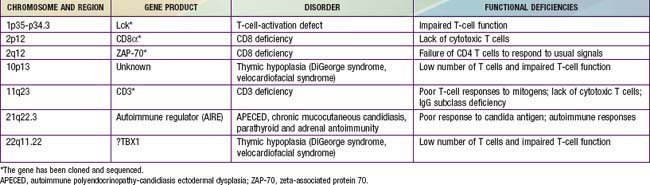
Thymic Hypoplasia (DIgeorge Syndrome)
Thymic hypoplasia results from dysmorphogenesis of the 3rd and 4th pharyngeal pouches during early embryogenesis, leading to hypoplasia or aplasia of the thymus and parathyroid glands. Other structures forming at the same age are also frequently affected, resulting in anomalies of the great vessels (right-sided aortic arch), esophageal atresia, bifid uvula, congenital heart disease (conotruncal, atrial, and ventricular septal defects), a short philtrum of the upper lip, hypertelorism, an antimongoloid slant to the eyes, mandibular hypoplasia, and low-set, often notched ears (Chapters 76 and 102). The diagnosis is often first suggested by hypocalcemic seizures during the neonatal period.
Genetics and Pathogenesis
DiGeorge syndrome occurs in both males and females. Microdeletions of specific DNA sequences from chromosome 22q11.2, the DiGeorge chromosomal region (DGCR), are found in a majority of cases. Several candidate genes have been identified in this region. A T-box transcription family member, TBX1, has been implicated as an etiology for most of the major signs of DGS. There appears to be an excess of 22q11.2 deletions of maternal origin. Polymerase chain reaction (PCR)-based genotyping using microsatellite DNA markers located within the commonly deleted region permits rapid detection of such microdeletions. Conotruncal heart defects and 22q deletions are observed in DiGeorge syndrome, velocardiofacial syndrome (VCFS), and conotruncal anomaly face syndrome (CTAFS). The CATCH 22 syndrome (cardiac, abnormal facies, thymic hypoplasia, cleft palate, hypocalcemia) includes the broad clinical spectrum of conditions with 22q11.2 deletions. Other deletions associated with DiGeorge and velocardiofacial syndromes have been identified on chromosome 10p13 (Chapter 76).



