Chapter 396 Primary Ciliary Dyskinesia (Immotile Cilia Syndrome)
Normal Ciliary Ultrastructure and Function
The upper and lower respiratory tracts are continuously exposed to inhaled pathogens, and local defenses have evolved to protect the airway. The respiratory epithelium in the nasopharynx, middle ear, paranasal sinuses, and larger airways are lined by a ciliated, pseudostratified columnar epithelium that is essential for mucociliary clearance (Fig. 396-1). Motile cilia are hairlike organelles that move fluids, mucus, and inhaled particulates vectorially from conducting airways, paranasal sinuses, and eustachian tubes.
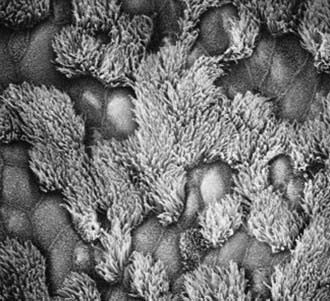
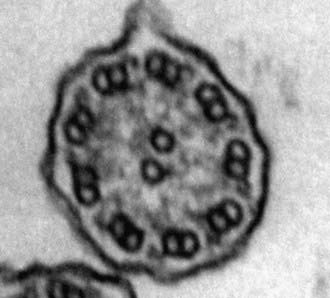
A mature ciliated epithelial cell has approximately 200 uniform motile cilia that are functionally and anatomically oriented in the same direction, moving with intracellular and intercellular synchrony. Anchored by a basal body to the apical cytoplasm and extending from the cell surface into the extracellular space, each cilium is a complex, specialized structure, composed of roughly 250 proteins. It contains a cylinder of microtubule doublets, arranged around a central pair of microtubules (Fig. 396-2), the characteristic “9+2” arrangement as viewed by cross-sectional views on electron microscopy. Multiple, different adenosine triphosphatases (ATPases), called dyneins, serve as “motors” of the cilium. Attached to the microtubules as distinct inner and outer dynein arms, dyneins cleave ATP to promote microtubule sliding, which is converted into bending. Nexin links and radial spokes act to restrict the degree of sliding between microtubules, allowing the cilium to bend.
A third distinct classification of cilia also exists, but only during embryonic development. These nodal cilia have a “9+0” microtubule arrangement similar to that of primary cilia, but they exhibit rotational movement, resulting in leftward flow of extracellular fluid that establishes body sidedness. Nodal cilia defects result in left-right body orientation abnormalities, such as situs ambiguus (Chapter 425.11).



