Preoperative and Postoperative Care of the Infant with Critical Congenital Heart Disease
John M. Costello
Wayne H. Franklin
INTRODUCTION
The contemporary strategy of early, complete repair for congenital heart defects originated in the 1970s, as evidenced by the first report in 1976 of complete anatomic correction of d-transposition of the great arteries (d-TGA) with ventricular septal defect (VSD) in early infancy (1). Aided by advances in surgical technique and equipment, myocardial protection, cardiac anesthesia and perioperative care, the strategy of early primary repair is now applied to many other complex congenital heart lesions, such as tetralogy of Fallot and truncus arteriosus (2,3).Furthermore, infants with certain lesions that were considered before the 1980s to be inoperable, such as hypoplastic left heart syndrome (HLHS), currently undergo staged surgical palliation with constantly improving outcomes (4). The first stage of surgical palliation for infants with single ventricle physiology usually must be performed in the neonatal period to minimize early morbidity and mortality, and to optimize the cardiovascular physiology for the subsequent Fontan operation. Early repair or palliation (for single ventricle lesions) benefits infants because prolonged preoperative periods of cyanosis (See Color Plate) and heart failure, and the many associated complications are minimized (5,6,7). These potential associated complications include impairment of cognitive function as a result of chronic hypoxemia, central nervous system embolic events, and the development of pulmonary vascular obstructive disease. Postoperative complications including pulmonary hypertensive crises are also reduced using a strategy of early surgical intervention (8). The incidence of vascular complications and infection related to central lines in infants requiring prolonged hospitalization prior to cardiac repair or palliation will also be minimized.
As a result of this early intervention strategy, approximately one-third of all patients with congenital heart defects undergo primary repair or palliation in early infancy. Due to the immaturity of many organ systems, these neonates possess limited physiologic reserve. They have limited ability to increase stroke volume, limited functional residual capacity in the lung, limited fat and carbohydrate reserves, and a limited ability to regulate temperature. Drug metabolism is altered by hepatic and renal immaturity, and total body water content. All of these factors complicate the care of infants in the perioperative period, and some have prolonged intensive care unit (ICU) stays, presenting a challenge for physicians involved with their management.
A variety of pediatric personnel with expertise in pediatric cardiology are involved in the day-to-day management of these patients. These personnel include neonatologists, intensivists, cardiologists, cardiac surgeons, anesthesiologists, nurses, and respiratory therapists. Because of the complexity inherent to this field, and to ensure an optimal outcome for each patient, seamless communication of pertinent anatomic, pathophysiologic, and operational/interventional details is required across all specialties.
This chapter will complement Chapter 33 by providing an overview of the key issues, concepts, and strategies pertaining to the care of infants with critical congenital heart
disease. We have allowed some overlap of information between chapters in the interest of clarity. The expected presentation and clinical course of specific congenital heart lesions will be discussed, and the general principles that are widely applied to patient management will be reviewed. This chapter will not provide a comprehensive description of every congenital heart defect and associated variants that may present in infancy. The reader must appropriately apply the concepts and principles discussed in the following pages to each unique patient. The first section will cover general preoperative issues, and the presentation, physiology and management of common types of critical congenital heart disease. The second section will provide an overview of cardiopulmonary bypass (CPB) and its sequelae, followed by a lesion specific discussion of operative intervention, postoperative physiology and common complications.
disease. We have allowed some overlap of information between chapters in the interest of clarity. The expected presentation and clinical course of specific congenital heart lesions will be discussed, and the general principles that are widely applied to patient management will be reviewed. This chapter will not provide a comprehensive description of every congenital heart defect and associated variants that may present in infancy. The reader must appropriately apply the concepts and principles discussed in the following pages to each unique patient. The first section will cover general preoperative issues, and the presentation, physiology and management of common types of critical congenital heart disease. The second section will provide an overview of cardiopulmonary bypass (CPB) and its sequelae, followed by a lesion specific discussion of operative intervention, postoperative physiology and common complications.
PREOPERATIVE CARE
Fetal Diagnosis
Fetal echocardiography plays an important role in the management of critical congenital heart disease for a variety of reasons (9). The identification of severe congenital heart disease during the second trimester allows time for family counseling. Fetuses that are likely to require surgical or transcatheter intervention in the neonatal period should ideally be delivered at an institution with a level II or III nursery that is located at or in close proximity to a tertiary care pediatric cardiology center. In the absence of nonimmune hydrops fetalis, fetuses with known critical congenital heart disease should be delivered at term to minimize the potential complications of prematurity, such as respiratory distress syndrome, intraventricular hemorrhage and necrotizing enterocolitis (NEC). As discussed below, infants with unrecognized critical congenital heart disease may develop severe cyanosis (See Color Plate), shock, myocardial dysfunction and end organ damage. Information from the fetal echocardiogram thus guides planning for initial stabilization upon delivery, such as the need for a prostaglandin E1 (PGE1) infusion. Prenatal diagnosis is associated with reduced preoperative morbidity and mortality in large series of neonates with HLHS and TGA (10,11,12,13). Management strategies may be developed in advance for the uncommon patient with congenital heart disease who may become critically ill in the first few minutes of life, such as some neonates with absent pulmonary valve syndrome, or HLHS with an intact atrial septum. The impact of in utero diagnosis of critical congenital heart disease on postoperative mortality is controversial. Some centers have reported lower postoperative mortality in patients with a fetal diagnosis of HLHS or d-TGA, whereas others found no such impact (12,13,14,15,16).
TABLE 34-1 RISK FACTORS FOR CONGENITAL HEART DISEASE THAT WARRANT FETAL ECHOCARDIOGRAPHY (SEE ALSO TABLE 33-6) | ||||||
|---|---|---|---|---|---|---|
| ||||||
Known risk factors for congenital heart disease that warrant referral to a pediatric cardiologist for fetal echocardiography are listed in Table 34-1 (17). Even if these screening guidelines are followed, the majority of patients with congenital heart disease will not be detected until after birth. Fetal echocardiography has several known limitations, most notably sub-optimal windows in mothers with large body habitus, and difficulty with precise imaging of extracardiac vessels. Two serious congenital heart lesions that may not be always appreciated with fetal echocardiography deserve comment. Isolated coarctation of the aorta may be difficult to definitively diagnose or exclude in utero as a result of the obligatory presence of the ductus arteriosus (18). Total anomalous pulmonary venous return may also be unrecognized. Thus, any diagnosis of fetal congenital heart disease must be confirmed, and the anatomic details clarified, as soon as possible after birth with a transthoracic echocardiogram.
Routine screening fetal ultrasound studies, which are interpreted by obstetricians, are usually limited to the demonstration of a heart with four chambers. Doppler assessment of blood flow patterns and valvar function is not usually performed, and data regarding longitudinal growth of the ventricular chamber sizes are not available when a single study is performed in the second trimester. Thus, a history of a normal fetal ultrasound does not exclude the possibility of critical congenital heart disease.
Transitional Circulation
An appreciation of the fetal-placental circulation, and the normal transition from fetal to newborn circulation, is required to understand the timing and presentation of symptoms in the neonate with critical heart disease (19). In the placenta, gas, nutrient, and waste exchange occur between the fetal and maternal circulations. Oxygenated blood then returns from the placenta to the fetus through the umbilical vein and partially bypasses the liver, via the ductus venosus, into the inferior vena cava. The oxygenated blood is preferentially shunted through the foramen ovale to the left atrium (20). The oxygen-rich blood thenenters the left ventricle, from where it is pumped out the aorta tosupply the coronary circulation and the brain. Deoxygenated blood from the superior vena cava and distal inferior vena cava preferentially enters the right ventricle, and is then pumped to the pulmonary artery. This preferential blood flow phenomenon is known as streaming. Due to the high pulmonary vascular resistance in utero, the majority of the deoxygenated pulmonary artery blood bypasses the lungs, and flows through the ductus arteriosus into the descending aorta, supplying the lower body and placenta.
An elegant series of events occur when the umbilical cord is clamped immediately after birth. The ductus venosus functionally closes in large part as a result of decreased flow from the placenta (21). The low resistance placental circulation is gone and thus systemic vascular resistance rises, although pulmonary vascular resistance falls as a result of mechanical expansion of the lungs with respiration and higher oxygen tension (22,23). Blood ejected from the right ventricle now perfuses the lungs rather than entering the ductus arteriosus, and the increased blood returning to the left atrium leads to functional closure of the foramen ovale. The increased oxygen tension also contributes to functional closure of the ductus arteriosus.
When the aforementioned events, in particular the fall in pulmonary vascular resistance and closure of the ductus arteriosus, occur in the setting of critical congenital heart disease, a physiologic state of either inadequate systemic or pulmonary blood flow exists which, if unrecognized, may progress to multi-organ system failure and death.
Presentation of Critical Congenital Heart Disease
Despite the broad spectrum of congenital heart lesions, neonates with critical congenital heart disease undetected in utero present in a limited number of ways: with cyanosis (inadequate pulmonary blood flow), shock (inadequate systemic blood flow) or some combination of these physiologic states. Older neonates and infants with significant left to right shunting at the ventricular or great artery level may present with congestive heart failure. Critical congenital heart disease may be detected prior to the onset of symptoms as a result of abnormal findings on cardiovascular examination or during echocardiographic screening performed in neonates with known chromosomal abnormalities or noncardiac congenital malformations.
Critical coarctation of the aorta, critical aortic valve stenosis, and HLHS are frequently encountered lesions that present in the neonatal period with symptomatic obstruction of the left ventricular outflow tract. In each case, constriction or closure of the ductus arteriosus leads to inadequate systemic blood flow. These patients will typically present within the first week or two of life with feeding difficulties, tachypnea, poor perfusion and metabolic acidosis, a constellation of findings that may be mistaken for septic shock. Due to low systemic cardiac output, findings such as pathologic heart murmurs may not be present and accurate four-extremity blood pressure measurements may be difficult to obtain. Diminished or absent pulses in the lower extremities and an increased right ventricular impulse are more reliable findings on physical examination, and a high index of suspicion is required to make a timely diagnosis.
Critical pulmonary valve stenosis, severe tetralogy of Fallot, and pulmonary atresia are examples of lesions with right ventricular outflow tract obstruction that manifest with progressive cyanosis in the newborn period as constriction of the ductus arteriosus limits pulmonary blood flow.
Lesions with complete intracardiac mixing of the systemic venous and pulmonary venous circulations, such as tricuspid atresia, truncus arteriosus, or complex single ventricles usually have variable degrees of obstruction to either the systemic or pulmonary circulations, depending upon the anatomic details of the individual lesion. Less commonly, they have no obstruction to either systemic or pulmonary blood flow, leading to mild hypoxemia (less commonly clinical cyanosis) but substantial heart failure that presents later in the neonatal period or during early infancy.
d-TGA is a common cyanotic lesion with the unique physiology of systemic and pulmonary circulations that run in parallel. Infants with d-TGA, an intact ventricular septum, and no significant outflow tract obstruction usually present with cyanosis soon after birth. Those with a significant VSD have less desaturation and can present later with congestive heart failure.
The neonate with nonimmune hydrops fetalis may have an underlying cardiac problem. The differential diagnosis includes serious congenital heart disease (particularly with significant atrioventricular valve regurgitation), obstructive cardiac tumors, congenital myocarditis, tachyarrhythmias and bradycardia.
Initial Evaluation and Stabilization of the Neonate with Suspected Congenital Heart Disease
A careful history and cardiovascular examination will often reveal abnormal findings in the infant with critical congenital heart disease, even before the onset of symptoms. Review of maternal records may identify a known risk factor for congenital heart disease (Table 34-1) (17). The course of labor
should be reviewed, and factors that suggest acute infection, respiratory distress syndrome, or meconium aspiration should be noted, as they may indicate a primary respiratory cause for hypoxemia. The physical examination focuses on the detection of dysmorphic features, cyanosis, an abnormal precordium, abnormal femoral and peripheral pulses, and/or a pathologic heart murmur. Measurement of blood pressure in all four extremities is useful if an arm-leg gradient of greater than 10 mm Hg is detected, but in the setting of a large patent ductus arteriosus, the pressures may be equal despite the presence of a critical aortic coarctation or interrupted aortic arch. Although respiratory distress may be present in neonates with critical congenital heart disease or pulmonary problems, those with heart disease typically have a shallow rapid respiratory pattern (“quiet tachypnea”), as opposed to the more labored breathing pattern of neonates with a primary pulmonary process.
should be reviewed, and factors that suggest acute infection, respiratory distress syndrome, or meconium aspiration should be noted, as they may indicate a primary respiratory cause for hypoxemia. The physical examination focuses on the detection of dysmorphic features, cyanosis, an abnormal precordium, abnormal femoral and peripheral pulses, and/or a pathologic heart murmur. Measurement of blood pressure in all four extremities is useful if an arm-leg gradient of greater than 10 mm Hg is detected, but in the setting of a large patent ductus arteriosus, the pressures may be equal despite the presence of a critical aortic coarctation or interrupted aortic arch. Although respiratory distress may be present in neonates with critical congenital heart disease or pulmonary problems, those with heart disease typically have a shallow rapid respiratory pattern (“quiet tachypnea”), as opposed to the more labored breathing pattern of neonates with a primary pulmonary process.
The term differential cyanosis refers to a condition in which the lower body is visibly more cyanotic than the upper body as a result of right to left shunting at the ductal level. The presence of differential cyanosis, occasionally seen with severe pulmonary hypertension, may also be indicative of aortic arch obstruction or coarctation of the aorta. In neonates with d-TGA, reverse differential cyanosis may be present if aortic arch obstruction or severe pulmonary hypertension also exists. In such patients, oxygenated blood is ejected from the left ventricle to the pulmonary artery, and then passes through the patent ductus arteriosus to the descending aorta.
In newborns that present with cyanosis, a right radial arterial blood gas analysis on room air and 100% inspired oxygen (the “hyperoxia test”) is useful for discriminating those with cyanotic heart disease from those with pulmonary disease. The partial pressure of carbon dioxide (pCO2) is typically mildly decreased in newborns with cardiac disease and mildly elevated in those with pulmonary disease. The arterial oxygen pressure (PaO2) is often between 25 to 40 mm Hg on room air in both patient groups. In 100% FiO2, the PaO2 will usually rise to more than 100 mm Hg in patients with pulmonary disease, provided that significant pulmonary artery hypertension is not present. The PaO2 will remain unchanged or only increase slightly in most neonates with cyanotic heart disease. There are limitations to the hyperoxia test. Patients with left sided obstructive lesions (e.g., HLHS, critical coarctation of the aorta, critical aortic valve stenosis, interrupted aortic arch) may have a high PaO2 (>60 mm Hg) in any extremity or a very high PaO2 (>150 mm Hg) in the right arm with certain anatomic variants. Pulse oximetry should not be used as a substitute for arterial blood gas analysis when performing the hyperoxia test. For example, a neonate receiving 100% fraction of inspired oxygen (FiO2) may have a pulse oximetry reading of 100%, with a PaO2 of 80 mm Hg (suggestive of cyanotic heart disease), or a PaO2 more than 100 mm Hg (suggestive of pulmonary disease). Potential complications exist when performing the hyperoxia test. For example, if the test is continued for a prolonged period of time in neonates with left sided obstructive lesions, pulmonary vascular resistance may drop further, and the ductus arteriosus may begin to close, leading to clinical deterioration.
Interpretation of a chest radiograph (CXR) and 12-lead electrocardiogram (ECG) may provide insight to the underlying cardiac diagnosis in cyanotic newborns. For example, cyanotic newborns with normal heart size, normal to increased pulmonary arterial markings on CXR, and a normal ECG may have TGA. Cyanotic newborns with normal heart size and decreased pulmonary blood flow on CXR are stratified by the ECG findings: those with right axis deviation (90°-180°) on ECG may have tetralogy of Fallot; those with a normal adult axis (10°-90°) on ECG may have pulmonary atresia with an intact ventricular septum; and those with an ECG with left axis deviation (0° to -90°) and left ventricular hypertrophy may have tricuspid atresia. Infants with a normal to small heart size and increased pulmonary vascular markings on CXR may have total anomalous pulmonary venous return, and those with a large heart with increased pulmonary vascular markings may have truncus arteriosus, or a complex single ventricle lesion with unobstructed pulmonary blood flow. Neonates with a very large heart and decreased pulmonary arterial markings may have Ebstein’s anomaly of the tricuspid valve.
If the aforementioned evaluation is consistent with the presence of critical congenital heart disease, it is reasonable to stabilize the infant, including the initiation of a PGE1 infusion in some cases, and then arrange transport to a pediatric cardiology center in which the initial echocardiogram can be performed. A PGE1 infusion should be initiated in neonates who present with shock, cyanosis with a minimal increase in PaO2 during the hyperoxia test, or a fetal diagnosis of ductal dependent congenital heart disease. An echocardiogram may be performed at the presenting institution if the presence of critical congenital heart disease is in doubt, or to guide initial stabilization if the transport time will be prolonged. The echocardiographer and the sonographer must have experience with congenital heart lesions, otherwise false negative and false positive evaluations may occur (24).
The introduction of prostaglandin infusions to maintain ductal patency in the late 1970s represented a major advance for infants with critical congenital heart disease (25,26). While receiving PGE1, infants can be safely transported over long distances to congenital heart centers. Cardiac surgery can be postponed while infections are treated and other major congenital anomalies identified. Patients who present in shock can be medically managed for several days, allowing time for recovery of end-organ function prior to surgery. Detailed cardiac diagnostic information can be obtained using echocardiography, cardiac catheterization, spiral computed tomography (CT), magnetic resonance imaging (MRI), or electron beam tomography (EBT) to allow optimal planning for intervention.
PGE1 can be administered through a peripheral intravenous line, although many practitioners prefer to use central venous access to ensure uninterrupted medication delivery. A wide range of dosing of PGE1 is used depending on the clinical state of the patient. Generally a PGE1 infusion
of 0.1 mcg/kg/min is used when the ductus arteriosus is severely constricted or functionally closed and a state of shock or severe cyanosis exists. Lower doses (0.01- 0.05 mcg/kg/min) will maintain ductal patency in neonates with presumed ductal dependent systemic or pulmonary circulation who are otherwise stable (27).
of 0.1 mcg/kg/min is used when the ductus arteriosus is severely constricted or functionally closed and a state of shock or severe cyanosis exists. Lower doses (0.01- 0.05 mcg/kg/min) will maintain ductal patency in neonates with presumed ductal dependent systemic or pulmonary circulation who are otherwise stable (27).
The most troublesome side effect of PGE1 infusion is apnea, which occurs in approximately one-third of neonates (27,28). Tracheal intubation and mechanical ventilation are usually indicated when PGE1 is started for neonates who present with severe cyanosis or shock and/or require transfer to another institution. Lower doses of PGE1 are associated with fewer side effects, particularly apnea (27,29). Patients who are stable upon presentation and are started on a low dose of PGE1 may be less likely to develop apnea, and their respiratory status may be observed without mechanical ventilation. This strategy may be used in patients who require transfer to another institution, provided that members of the transport team are experienced and prepared to intubate the patient’s trachea should the need arise. Preliminary data suggest that aminophylline may be prescribed to minimize the occurrence of apnea in neonates requiring PGE1 infusion (30). PGE1 is a potent vasodilator, and hypotension may occur following the initiation of the drug, particularly if narcotics are also administered to facilitate procedures. This problem should be anticipated, and the hypotension usually resolves with a dose reduction of PGE1 and volume administration. Other common side effects are listed in Table 34-2 (27,28). Although unusual, clinical deterioration may occur following the initiation of a PGE1 infusion, and in these instances, obstruction to pulmonary venous return (e.g., obstructed total anomalous pulmonary venous return) or left atrial egress (e.g., TGA with intact ventricular septum and a restrictive atrial communication) may be present. These infants require emergent surgical or catheter intervention. Some neonates have congenital absence of the ductus arteriosus (such as those with tetralogy of Fallot with absent pulmonary valve syndrome) or some infants with pulmonary atresia, ventricular septal defect, and major aortopulmonary collateral arteries. A PGE1 infusion in these patients may worsen cyanosis by lowering systemic vascular resistance and thereby decreasing pulmonary blood flow.
When endotracheal intubation is indicated for a neonate with critical congenital heart disease, the use of medications should be considered to blunt the stress and vagal responses to laryngoscopy, decrease oxygen consumption and thus improve oxygen delivery, and provide pharmacologic paralysis to facilitate the procedure. The choice of specific medications depends on the clinical scenario and the airway skills of the clinician. As mechanical ventilation is initiated, care must be taken in many instances to avoid excessive use of supplemental oxygen and hyperventilation. These maneuvers may lower pulmonary vascular resistance, thereby increasing the ratio of pulmonary to systemic blood flow (Qp/Qs), and “stealing” blood flow from the systemic circulation. This is discussed in greater detail in the HLHS section below.
TABLE 34-2 SIDE EFFECTS OF PROSTAGLANDIN E1 INFUSION | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||
Stable intravenous access is required for all infants with critical congenital heart disease. The need for arterial and central venous access should be determined on a case-by case basis, based upon the heart defect, clinical presentation and anticipated preoperative course. Central access may be needed to allow for medication delivery, frequent blood sampling, and the administration of parenteral nutrition. The most accessible vessels in the newborn are the umbilical arteries and veins. The use of umbilical vessels for preoperative stabilization leaves other vessels available for future cardiac catheterizations and surgical procedures. If umbilical lines cannot be placed, the radial or femoral arteries and the femoral veins may be accessed. Some clinicians prefer to avoid placing central venous catheters in the internal jugular or subclavian veins in patients with single ventricle physiology to prevent scarring and clot formation, which may impede venous return following the bi-directional Glenn/hemi-Fontan and Fontan operations.
Adequate cardiac output and oxygen delivery usually exist in infants with critical congenital heart disease following initial stabilization and infusion of PGE1. Infants who demonstrate signs of inadequate cardiac output may benefit from inotropic infusions, but should be critically reassessed to ensure that the PGE1 is being delivered correctly, that appropriate oxygenation and ventilation strategies are being employed to prevent pulmonary over-circulation, and that noncardiac etiologies for shock are not present, such as pneumothorax or sepsis. Fetal-maternal hemorrhage, twin-twin transfusion, and blood loss at delivery from delayed cord clamping are examples of perinatal causes of anemia that may necessitate transfusion of packed red blood cells soon after delivery. Most patients with ductal dependent congenital heart disease require a hemoglobin level of 13 to
15 gm/dL. Attention also needs to be given to acid-base status, and blood glucose and calcium levels.
15 gm/dL. Attention also needs to be given to acid-base status, and blood glucose and calcium levels.
Bacterial pneumonia and sepsis are often considered when an infant presents with cyanosis and shock, and empiric antibiotics may be prescribed before a diagnosis of critical congenital heart disease is confirmed. If no source of bacterial infection is identified, the antibiotics may be discontinued after a 48-hour period. Although it has been suggested that the use of PGE1 increases the risk of bacterial infection, there is no published evidence to support this concept.
Neonates with critical congenital heart disease require transport to congenital heart centers for further evaluation and intervention. Experienced pediatric transport teams are used when available, and close communication is required with the intensivist and pediatric cardiologist at the congenital heart center throughout the transport process. During transport, care must be taken to maintain a normal patient temperature as hypothermia will increase systemic vascular resistance and may be poorly tolerated. The transport team members must understand that normal ventilation and the avoidance of excessive supplemental oxygen are important to maintain a balanced circulation in many infants with ductal dependent systemic or pulmonary blood flow.
Evaluation at the Congenital Heart Center
Upon arrival to the tertiary care center, the intensivist and nursing staff should obtain a detailed report from the transport team. Historical features that are useful to direct the diagnostic evaluation and initial management include the results of fetal echocardiogram(s), complications of the pregnancy, family history, estimated gestational age of the fetus, severity of cyanosis or shock prior to initial resuscitation, presence of risk factors for infection, and suspected noncardiac congenital anomalies.
A detailed physical examination includes a review of recent and current vital signs. Heart rates are usually within the normal range for age. Four-extremity blood measurements should be obtained. Pulse oximetry results are interpreted in the context of the patient’s physiology. The presence of dysmorphic facial features may suggest a specific genetic syndrome. The cardiac examination begins with inspection and palpation of the precordium. An increased right ventricular impulse is often present with significant left ventricular outflow tract obstruction and/or pulmonary hypertension. Auscultation is performed with attention to splitting and quality of the second heart sound, systolic ejection clicks, and the presence of pathologic systolic or diastolic murmurs. The span of the liver is determined, and the quality of the peripheral pulses and perfusion are noted.
The ECG and CXR are reviewed with attention to features that may suggest an underlying cardiac diagnosis as described above. The CXR should also be reviewed to ensure that any tubes or lines placed prior to transport have not migrated. Cardiopulmonary instability in the infant with critical congenital heart disease is often as a result of noncardiac issues, such as respiratory distress syndrome, tension pneumothorax, or endotracheal tube plugging or malposition.
Even if an echocardiogram has been performed at the referring institution, a complete echocardiogram is obtained upon arrival to the congenital heart center to confirm the diagnosis and clarify anatomic and physiologic details. Complete diagnostic information is obtained in the vast majority of patients with critical congenital heart disease using transthoracic echocardiogram, eliminating the need for diagnostic cardiac catheterization (31,32). Occasionally a critically ill infant with congenital heart disease will not tolerate a prolonged echocardiogram, as a result of temperature instability related to exposure, or hypotension related to abdominal compression during subcostal imaging.
Limited indications exist for diagnostic cardiac catheterization in the current era. For example, infants with pulmonary atresia, VSD and major aortopulmonary collateral arteries (MAPCAs) usually require catheterization to define all sources of pulmonary arterial blood flow. Coronary anatomy can usually be clarified by echocardiography, but occasionally angiography is required, as is the case in some infants with tetralogy of Fallot, pulmonary atresia with intact ventricular septum, or TGA (33). Hemodynamic data, such as pulmonary vascular resistance, may need to be obtained by cardiac catheterization in selected older infants prior to a definitive procedure. Cardiac catheterization is more commonly indicated to perform interventional procedures. For example, neonates with TGA and intact ventricular septum may require enlargement of the atrial communication, and those with HLHS and intact atrial septum require emergent creation of an adequate atrial septal defect (ASD) to allow egress of pulmonary venous blood from the left atrium into the right atrium (34,35). Balloon valvuloplasty of critical aortic or pulmonary valve stenosis is commonly performed in many centers. Cardiac MRI, spiral CT, and electron beam tomography are evolving tools that are currently available in selected centers and are most useful for clarifying anatomy of extracardiac vessels (36,37).
Evaluation of other organ systems is indicated in infants with critical congenital heart disease prior to intervention. Basic laboratory studies are obtained to evaluate acid-base status, oxygenation and ventilation, and the hepatic, renal and hematologic systems. Recovery of end-organ function in infants who present in shock is to be expected in nearly all cases following appropriate resuscitation and stabilization. Infants who are premature or who present with severe cyanosis or shock should have a head ultrasound performed to evaluate for intracranial hemorrhage. If not clear by echocardiography, an abdominal ultrasound is indicated in patients with heart disease associated with heterotaxy syndrome to determine the sidedness of the liver and spleen. An upper gastrointestinal series may be needed in such infants to evaluate for malrotation. Any suspected hematologic issues, such as thrombocytopenia or a bleeding diathesis, should be thoroughly evaluated prior to intervention. Other major birth defects may be seen in up
to 25% of patients with significant congenital heart disease. For example, infants with conotruncal heart defects, such as tetralogy of Fallot, pulmonary atresia with VSD, interrupted aortic arch, and truncus arteriosus have an increased risk compared with the general population of having an associated oral cleft, omphalocele, trachoesophageal fistula, or imperforate anus (38). There is an increased incidence of renal anomalies in infants with congenital heart disease who have other major congenital anomalies, and a screening renal ultrasound may be indicated in this subset of patients (39).
to 25% of patients with significant congenital heart disease. For example, infants with conotruncal heart defects, such as tetralogy of Fallot, pulmonary atresia with VSD, interrupted aortic arch, and truncus arteriosus have an increased risk compared with the general population of having an associated oral cleft, omphalocele, trachoesophageal fistula, or imperforate anus (38). There is an increased incidence of renal anomalies in infants with congenital heart disease who have other major congenital anomalies, and a screening renal ultrasound may be indicated in this subset of patients (39).
A chromosomal abnormality or easily definable genetic syndrome may be identified in approximately 20% of infants with congenital heart disease. Table 34-3 lists selected common chromosomal abnormalities and genetic syndromes, and the congenital heart lesions with which they are associated (40,41,42).
Preoperative Supportive Care: General Principles
Cardiopulmonary Management
Infants with ductal dependent congenital heart disease require a “balanced” circulation to achieve adequate systemic and pulmonary blood flow. Those who present with severe cyanosis or shock, or require prolonged interhospital transport are tracheally intubated and mechanical ventilation is provided. Great care must be taken throughout the preoperative period to ensure that appropriate ventilation and oxygenation strategies are used to prevent an iatrogenic imbalance between systemic and pulmonary blood flow. In particular, in patients with ductal dependent systemic blood flow (e.g., HLHS or interrupted aortic arch), hyperventilation and provision of supplemental oxygen will decrease pulmonary vascular resistance, thereby promoting increased pulmonary to systemic flow ratio (Qp/Qs), and “steal” from the systemic and coronary circulations. Such patients will have inappropriately high systemic oxygen saturations (greater than 90%), poor systemic perfusion and are at risk of developing myocardial dysfunction as a result of volume overload and coronary ischemia, renal insufficiency, and/or NEC. Interventions that may be employed to increase pulmonary vascular resistance and improve the balance between systemic and pulmonary circulations, including subambiant FiO2 and hypoventilation, are discussed below in the section on preoperative management of HLHS. Neonates with left ventricular outflow tract obstruction who are stable at presentation may be allowed to spontaneously breathe while awaiting surgical intervention, with close monitoring of their cardiopulmonary status. These neonates will typically have relatively high systemic oxygen saturations (greater than 90%) and “quiet tachypnea,” but will maintain adequate peripheral pulses, urine output, and low
lactate levels. Diuretics and low dose inotropic support are occasionally needed to alleviate pulmonary edema and to support the volume-loaded ventricular myocardium. In neonates with right ventricular outflow tract obstruction, the ductus arteriosus is often somewhat stenotic and takes a tortuous course from the aorta to the pulmonary artery, related to the in utero blood flow pattern, which typically will prevent torrential pulmonary blood flow. Thus marked pulmonary overcirculation is less of a clinical concern in ductal-dependent neonates with right ventricular outflow tract obstruction.
lactate levels. Diuretics and low dose inotropic support are occasionally needed to alleviate pulmonary edema and to support the volume-loaded ventricular myocardium. In neonates with right ventricular outflow tract obstruction, the ductus arteriosus is often somewhat stenotic and takes a tortuous course from the aorta to the pulmonary artery, related to the in utero blood flow pattern, which typically will prevent torrential pulmonary blood flow. Thus marked pulmonary overcirculation is less of a clinical concern in ductal-dependent neonates with right ventricular outflow tract obstruction.
TABLE 34-3 SELECTED COMMON CHROMOSOMAL ABNORMALITIES AND GENETIC SYNDROMES ASSOCIATED WITH CONGENITAL HEART DISEASE (SEE ALSO TABLE 33-5) | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Inotropic support is occasionally necessary for neonates with congenital heart disease, particularly those with volume loaded single ventricles, but escalating inotropic or vasopressor requirements often signify a noncardiac problem or inadequate systemic cardiac output as a result of Qp/Qs imbalance. Inotropic agents are discussed in the postoperative section on low cardiac output.
Prematurity
The premature infant with critical congenital heart disease presents a significant challenge. Problems inherent to prematurity, including temperature instability, decreased nutritional reserve, intraventricular hemorrhage, respiratory distress syndrome, infection, and NEC, may develop during the pre- and postoperative course. In preterm infants with critical congenital heart disease, a management strategy of prolonged medical therapy to achieve weight gain prior to surgery is fraught with complications related to infection, heart failure and feeding intolerance (43,44). Advances in cardiopulmonary bypass techniques and miniaturization of surgical equipment allow for the safe conduct of open-heart surgery in these patients. Several reports depict the recent experience with palliative or definitive surgical intervention in premature infants with critical congenital heart disease. Reddy and associates reported the University of California at San Francisco experience with complete (two-ventricle) repair in 102 infants with significant congenital heart disease weighing less than 2,500 grams. These investigators found higher preoperative morbidity in the group of patients having delayed repair. The early and late mortalities were 10 and 8%, respectively, and no patient had postbypass intracranial hemorrhage (44). Investigators from the same center reported a series of 20 very low birth weight (LBW)(< 1,500 gm) infants with symptomatic congenital heart disease who underwent complete repair with only two deaths (10%) (45). Rossi and associates reported a series of infants weighing less than 2 kg, 73% of which were born prematurely. Hospital survival was 83%, and 24% of the patients had neurologic complications (46). Bacha and associates, in a study of 18 infants weighing less than 2 kg with coarctation of the aorta, found that repair can be accomplished with low operative mortality, but the residual or recurrent coarctation rate was 44% (47). Results for single ventricle palliation are less encouraging. The reported mortality for infants weighing less than 2.5 kg at the time of Norwood operation for HLHS or other single ventricle variants is 45% to 51% (48,49). Thus, although prematurity and LBW complicate the care of infants with significant heart disease, early intervention, including corrective surgery, can be performed with acceptable morbidity and mortality.
Nutrition
Although the enteral route is the preferred method for providing nutrition to infants in the intensive care unit, those with critical congenital heart disease awaiting intervention often have several pathophysiologic features that must be considered prior to initiation of enteral feedings. The combination of myocardial dysfunction, cyanosis and falling pulmonary vascular resistance, the later allowing increased diastolic runoff through the ductus arteriosus, may result in inadequate mesenteric oxygen delivery. In one large, retrospective study, approximately 3% of neonates admitted to a busy cardiac intensive care unit developed NEC (50). Independent risk factors for developing NEC were prematurity, a history of resuscitation from severe cyanosis or shock, HLHS, or the presence of diastolic runoff through the ductus arteriosus or other aorto-pulmonary connections (50). In part as a result of the absence of randomized studies, the use of enteral nutrition in infants with ductal-dependent congenital heart disease remains controversial and clinical practice varies widely. The risk-benefit ratio for providing enteral nutrition must be individually determined for each patient. One approach is to avoid enteral feeding in those infants who have one or more of the aforementioned risk factors. It is also reasonable to consider enteral feedings in those infants with an acceptable diastolic blood pressure despite the presence of a patent ductus arteriosus, provided that additional risk factors are not present.
Congenital Complete Heart Block
Congenital complete heart block may be associated with structural heart disease including atrioventricular canal defects and complex lesions with ventricular inversion. In neonates with normal intracardiac anatomy, there is a high incidence of maternal autoimmune disease, particularly systemic lupus erythematosus (51). Congenital complete heart block is diagnosed electrocardiographically when atrioventricular dissociation is present with an atrial rate that is appropriate for age and a slow ventricular rate. The diagnosis may be made in utero by fetal echocardiography and may be associated with hydrops fetalis. Dual chamber epicardial pacemaker placement is indicated when third degree congenital atrioventricular heart block is associated with a wide QRS escape rhythm, complex ventricular ectopy, or ventricular dysfunction. A pacemaker is also indicated in an infant with a ventricular rate less than 50 to 55 beats per minute, or with congenital heart disease and a ventricular rate less than 70 beats per minute (52).
PREOPERATIVE PHYSIOLOGY AND MANAGEMENT: SPECIFIC LESIONS
Right Ventricular Outflow Tract Obstruction
Critical Pulmonary Valve Stenosis
In neonates with critical pulmonary valve stenosis, the pulmonary valve leaflets are thickened and fused to a variable extent, and their mobility is decreased (Fig. 34-1, see also color plate). The pulmonary valve annulus is hypoplastic in severe cases. The pulmonary arteries are usually of normal size, but may be stenotic or dilated. The right ventricle is often hypertrophied and mildly hypoplastic. Significant tricuspid regurgitation, and a right to left atrial shunt are present (53). Infants with isolated critical pulmonary valve stenosis will develop significant cyanosis (See Color Plate) upon closure of the ductus arteriosus. Such infants are dependent on a PGE1 infusion to maintain ductal patency for adequate pulmonary blood flow. Although open surgical valvotomy was routinely performed in the past to relieve right ventricular outflow tract obstruction, most of these infants are candidates for a balloon valvuloplasty in the cardiac catheterization laboratory. Balloon valvuloplasty is successful in approximately 60% to 80% of cases (53,54,55,56,57,58). Neonates who fail an attempted balloon valvuloplasty may have smaller right heart structures, and are referred for a surgical valvotomy +/- a systemic to pulmonary artery shunt (59,60).
Tetralogy of Fallot
Tetralogy of Fallot, the most common cyanotic congenital heart lesion, is comprised of an anterior malalignment VSD in a heart with two ventricles and aortic to mitral fibrous continuity. Anatomic details that need to be clarified at the time of diagnosis include the severity and location of right ventricular outflow tract obstruction, the pulmonary artery anatomy, the presence of additional VSDs, the sidedness of the aortic arch (rightward in 25%), and the coronary artery anatomy. The left coronary artery may arise from the right coronary artery and cross the right ventricular outflow tract in 5% of cases. Complete anatomic information is usually obtainable by transthoracic echocardiogram (33). If uncertainty exists about the coronary artery anatomy, some surgeons may request a cardiac catheterization, as great care must be taken not to injure the coronary vessels during right ventricular outflow tract reconstruction. Chromosome 22q11 deletion is present in 15% of patients with tetralogy of Fallot, and those with a right aortic arch are at higher risk (41).
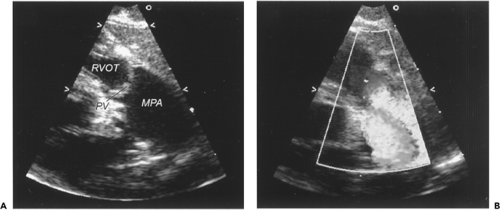 Figure 34-1 Critical pulmonary valve stenosis. A. Two-dimensional echocardiogram from the parasternal long axis view demonstrating thickened and doming pulmonary valve leaflets. Note the poststenotic dilation of the main pulmonary artery. B. Application of color Doppler during ventricular systole demonstrates a turbulent flow jet originating at the pulmonary valve leaflets and extending into the main pulmonary artery. MPA, main pulmonary artery; PV, pulmonary valve; RVOT, right ventricular outflow tract. (See color plate) |
In neonates with tetralogy of Fallot, a wide spectrum of presentation exists, depending in large part upon the degree of malalignment of the conal septum into the right ventricular outflow tract. Infants with a minimal degree of obstruction to pulmonary blood flow, commonly referred to as “pink tets,” are usually asymptomatic and fairly well oxygenated (systemic saturations > 85%) soon after birth. They are commonly observed in the hospital until the ductus arteriosus closes and then sent home to await surgical repair within the first 6 months of life. Occasionally a “pink tet” will mimic the pathophysiology of an infant with a large VSD and develop congestive heart failure within the first few months of life as pulmonary vascular resistance falls. More commonly these patients develop progressive right ventricular outflow tract obstruction and cyanosis. The exact timing of repair varies depending
upon several variables including the development of increasing cyanosis, hypercyanotic spells (“TET spells”), and institutional preference (2,61). Infants with tetralogy of Fallot and more severe right ventricular outflow tract obstruction or pulmonary atresia will develop excessive cyanosis (oxygen saturation < 75%-80%) upon closure of the ductus arteriosus. Such patients will require surgical intervention during the initial hospitalization. Either a complete repair or a systemic to pulmonary shunt is performed, depending on the details of the anatomy and surgeon preference (62).
upon several variables including the development of increasing cyanosis, hypercyanotic spells (“TET spells”), and institutional preference (2,61). Infants with tetralogy of Fallot and more severe right ventricular outflow tract obstruction or pulmonary atresia will develop excessive cyanosis (oxygen saturation < 75%-80%) upon closure of the ductus arteriosus. Such patients will require surgical intervention during the initial hospitalization. Either a complete repair or a systemic to pulmonary shunt is performed, depending on the details of the anatomy and surgeon preference (62).
Hypercyanotic episodes, or “TET spells,” are potentially life-threatening events notable for significant desaturation and irritability as a result of an acute decrease in pulmonary blood flow. The precise etiology of these spells is unclear but has been attributed to spasm of the infundibular conus, an imbalance between the systemic and pulmonary vascular resistances, increased systemic venous return, and/or tachycardia, all of which may lead to a progressive cycle of decreased pulmonary blood flow, increased cyanosis, and eventually metabolic acidosis. Although TOF spells can occur at any age, the incidence seems to increase after 4 to 6 months of age, which is one of the reasons that early complete repair before this age is commonly recommended. TET spells can be triggered by medical procedures including placement of intravenous catheters and cardiac catheterization. Infants with tetralogy of Fallot have a fixed component of obstruction to pulmonary blood flow, and thus will normally develop transient desaturation with agitation and feeding as a result of increased oxygen consumption and cardiac output. Clinical judgment is required to differentiate a true TET spell from these common and benign desaturation episodes. The absence of a murmur during a true TET spell, signifying a substantial reduction of blood flow across the right ventricular outflow tract, may be useful in this regard. Treatment strategies are implemented with the goal of decreasing patient agitation and heart rate, increasing systemic vascular resistance and pulmonary blood flow, and correcting metabolic acidosis (Table 34-4). Prompt surgical intervention is indicated once an infant has had a TET spell.
Absent pulmonary valve syndrome, identified by the presence of rudimentary pulmonary valve leaflets and free pulmonary insufficiency, is a relatively rare lesion, present in approximately 3% of all patients with tetralogy of Fallot. This lesion is most notable for in utero development of aneurysmal dilation of the pulmonary arteries, which may occur as a result of the lack of a ductus arteriosus or as a result of the pulsatile blood flow pattern across the right ventricular outflow tract. When compared with infants with typical tetralogy of Fallot, there may be a higher incidence of chromosome 22q11 deletion in those with absent pulmonary valve (63). A nearly pathognomonic to-and-fro (“sawing wood”) murmur is heard as a result of pulmonary annulus stenosis and free pulmonary insufficiency (Fig. 34-2, see also color plate). Such patients may have minimal if any respiratory symptoms and behave much like a “pink tet,” or present with severe problems with oxygenation and ventilation soon after birth as a result of compression of the bronchi by the dilated pulmonary arteries (64). Intubation, mechanical ventilation (with judicious use of positive end-expiratory pressure), and deep sedation may be life saving in neonates with symptomatic airway obstruction. Prone positioning may be advantageous as gravity will allow the pulmonary arteries to fall off the mainstem bronchi, thus permitting adequate gas exchange (65). Prompt surgical repair is indicated in symptomatic neonates.
TABLE 34-4 TREATMENT FOR HYPERCYANOTIC EPISODES (“TOF SPELLS”) | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Pulmonary Atresia, Ventricular Septal Defect, Major Aortopulmonary Collateral Arteries
Infants with pulmonary atresia, VSD and MAPCAs represent a challenging subset of infants with pulmonary atresia. This lesion is also referred to as tetralogy of Fallot, pulmonary atresia and MAPCAs. In addition to the anatomic features of tetralogy of Fallot as described above, the true pulmonary arteries are often diminutive or absent, and a variable number of aortopulmonary collaterals arise from the aorta or bracheocephalic vessels to supply pulmonary blood flow. Infants with this lesion will present with either cyanosis or congestive heart failure, depending upon the cardiac anatomy (66). The physical examination may be notable for the presence of a widely radiating continuous murmur produced by the aortopulmonary collateral blood flow. These infants are generally not dependent upon prostaglandin infusion unless a major aortopulmonary
collateral vessel arises from the ductus arteriosus. Cardiac catheterization is required to identify all sources of pulmonary blood flow. Surgical strategy must then be individualized depending upon the pulmonary artery and collateral vessel anatomy. True pulmonary arteries, MAPCAs, or both may supply blood to individual segments of the lungs. The ultimate goal is to encourage true pulmonary artery growth and incorporate as many arterial blood vessels and lung segments into the repair as possible to maximize the effective pulmonary arterial vascular bed, and thus minimize right ventricular hypertension following VSD closure. In situations in which dual blood supply to a lung segment exists, the MAPCAs are coil occluded in the cardiac catheterization laboratory or ligated at the time of surgery to eliminate left to right shunting. As with other conotruncal defects, these infants should undergo genetic testing for chromosome 22q11 deletion.
collateral vessel arises from the ductus arteriosus. Cardiac catheterization is required to identify all sources of pulmonary blood flow. Surgical strategy must then be individualized depending upon the pulmonary artery and collateral vessel anatomy. True pulmonary arteries, MAPCAs, or both may supply blood to individual segments of the lungs. The ultimate goal is to encourage true pulmonary artery growth and incorporate as many arterial blood vessels and lung segments into the repair as possible to maximize the effective pulmonary arterial vascular bed, and thus minimize right ventricular hypertension following VSD closure. In situations in which dual blood supply to a lung segment exists, the MAPCAs are coil occluded in the cardiac catheterization laboratory or ligated at the time of surgery to eliminate left to right shunting. As with other conotruncal defects, these infants should undergo genetic testing for chromosome 22q11 deletion.
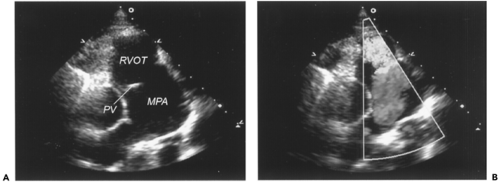 Figure 34-2 Tetralogy of Fallot with absent pulmonary valve. A. Two-dimensional echocardiogram from the parasternal long axis view demonstrating rudimentary pulmonary valve leaflets and aneurysmal dilation of the main pulmonary artery. B. Application of color Doppler during ventricular diastole revealing free regurgitation from the main pulmonary artery into the right ventricular outflow tract. MPA, main pulmonary artery; PV, pulmonary valve; RVOT, right ventricular outflow tract. (See color plate) |
Pulmonary Atresia with Intact Ventricular Septum
Pulmonary atresia is characterized by a membranous or plate-like obstruction of the right ventricular outflow tract at the level of the pulmonary valve, with variable degrees of hypoplasia of the tricuspid valve and right ventricle, and occasional coronary artery fistulous connections to the right ventricle. Systemic venous return to the right atrium flows through an obligatory atrial communication to the left atrium. Infants with pulmonary atresia and intact ventricular septum will manifest with significant cyanosis upon closure of the ductus arteriosus, and are thus dependent on prostaglandin infusion. Intervention in these patients depends on two key issues: the adequacy of the right ventricular size to support a two-ventricular circulation, and on the presence of “right ventricle dependent coronary circulation.” The right ventricle must have adequate size to support the entire right heart circulation if these patients are to be considered for a two-ventricle repair. The size of the tricuspid valve annulus plays an important role in the decision making process (67). Infants with pulmonary atresia and intact ventricular septum, particularly those with small tricuspid valve annulus size, may have significant fistula communications between the right ventricle and the coronary circulation, known as coronary-cameral fistulae, which may be identified by echocardiogram and further characterized by cardiac catheterization (68,69). If coronary stenoses exist such that a significant amount of coronary blood flow to the myocardium is derived from and dependent upon the coronary cameral fistulae, then “right ventricular dependent coronary circulation” is present. If the right ventricular outflow tract obstruction is relieved in an infant with “right ventricular dependent coronary circulation,” coronary perfusion pressure falls and the patient may develop a myocardial infarction. Thus, if the right ventricle is not of adequate size, or if “right ventricle dependent coronary circulation” exists, these patients are referred for a systemic to pulmonary artery shunt in anticipation of an eventual Fontan operation. If a complete two-ventricle repair is deemed appropriate, this may be performed as a primary operation in the neonatal period, although more commonly a systemic to pulmonary artery shunt and right ventricular outflow tract decompression is initially performed to encourage right ventricular growth, with complete repair in later infancy (70). Some infants with membranous pulmonary atresia and intact ventricular septum who meet criteria for a two ventricular repair may undergo perforation of the pulmonary valve using a stiff wire, a radiofrequency ablation catheter, or a laser catheter, followed by balloon valvuloplasty (54,58,71). In well-selected patients, the use of transcatheter intervention
may eliminate the need for surgical intervention. The decision to attempt transcatheter intervention is made considering patient size, co-morbidities, anatomic details, and institutional experience.
may eliminate the need for surgical intervention. The decision to attempt transcatheter intervention is made considering patient size, co-morbidities, anatomic details, and institutional experience.
Ebstein’s Anomaly of the Tricuspid Valve
Ebstein’s anomaly of the tricuspid valve is a malformation in which the septal and posterior leaflets of the tricuspid valve are displaced to a variable extent into the anatomic right ventricle, and the anterior leaflet, although not inferiorly displaced, is redundant. Tricuspid regurgitation of variable severity occurs (Fig. 34-3, see also Color Plate). One or more accessory connections may be present at the tricuspid valve annulus, creating the necessary substrate for atrio-ventricular reentrant tachycardia. The clinical presentation is usually one of right-sided congestive heart failure and arrhythmias in adolescence or early adulthood. A small subset of infants with Ebstein’s anomaly, those with severe tricuspid regurgitation, a large “atrialized” portion of the right ventricle, severe cardiomegaly, and pulmonary hypoplasia are critically ill soon after birth and have a high mortality (72). These neonates will present with severe cyanosis as a result of functional right ventricular outflow tract obstruction, and right to left shunting at the atrial level. Infants with significant cyanosis should be started on PGE1, and judicious use of positive end-expiratory pressure is required to re-expand the lungs, which are compressed by the generous heart size (73). Additionally, standard measures including inhaled nitric oxide should be employed to decrease pulmonary vascular resistance and cyanosis. In many of these patients, as pulmonary vascular resistance falls over time, forward flow increases across the right ventricular outflow tract, cyanosis decreases, and surgery can be deferred. For those neonates with refractory cyanosis, referral is made for either a primary complete repair, consisting of a reduction atrioplasty, fenestrated closure of the atrial septum and tricuspid valvuloplasty, or palliation toward the Fontan pathway starting with a systemic to pulmonary artery shunt (73,74,75).
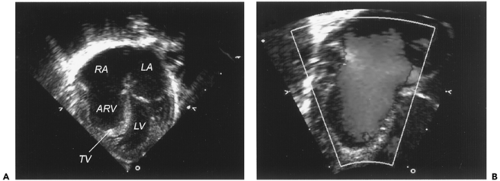 Figure 34-3 Ebstein’s anomaly of the tricuspid valve. A. Two-dimensional echocardiogram from the apical four-chamber view demonstrating significant displacement of the tricuspid valve leaflets into the right ventricle. B. During ventricular diastole, color Doppler demonstrating free retrograde flow across the tricuspid valve annulus. ARV, atrialized right ventricle; LA, left atrium; LV, left ventricle; RA, right atrium; TV, tricuspid valve. (See color plate) |
Left Ventricular Outflow Tract Obstruction
Critical Aortic Valve Stenosis
Isolated congenital aortic valve disease may present in a variety of ways, depending primarily upon the severity of stenosis. A bicuspid aortic valve without stenosis or insufficiency may be an incidental finding in an otherwise healthy patient. At the other extreme, newborns with critical aortic valve stenosis develop shock upon closure of the ductus arteriosus. The murmur is typically softer than expected as a result of the low cardiac output state, and the gradient estimated by echocardiography may not correlate with the severity of the stenosis. Initiation of PGE1 infusion allows blood from the right ventricle to provide systemic perfusion until an intervention can be performed (76). Once the infant is stabilized, echocardiographic information must be carefully reviewed to predict whether the left ventricle is of adequate size to sustain the systemic circulation following a two-ventricle repair (77). Infants thought to be suitable for a two-ventricle repair usually will be referred for a balloon aortic valvuloplasty; although a surgical aortic valvotomy is an acceptable option (78,79,80). If hypoplasia of multiple left sided structures suggests that the left ventricle will not be able to support the circulation, then a Norwood operation or heart transplantation may be considered.
Critical Coarctation of the Aorta
A coarctation of the aorta is present when a significant narrowing exists in the thoracic aorta just distal to the left
subclavian artery. A “posterior shelf” is present opposite the insertion site of the patent ductus arteriosus. Coarctation of the aorta may exist in isolation or can be associated with aortic arch hypoplasia, a VSD, or other complex intracardiac lesions. When present in isolation, coarctation of the aorta is “critical” when ductal dependent systemic blood flow exists, which represents only about 10% of cases. Neonates with isolated critical coarctation of the aorta also will present with shock if the diagnosis is not made before significant constriction of the ductus arteriosus occurs (Fig. 34-4). Prior to the development of shock, the diagnosis should be suspected by the detection of a brachial-femoral pulse discrepancy, which can be confirmed by measuring an arm-leg blood systolic pressure gradient. A systolic blood pressure gradient between the upper and lower extremities of greater than 10 mm Hg is clinically significant. In approximately 5% of infants with isolated coarctation of the aorta, the right subclavian artery will arise aberrantly from the descending thoracic aorta, distal to the coarctation. Thus it is important to palpate pulses and measure blood pressure in both upper extremities in all infants with suspected coarctation. Occasionally differential cyanosis will be present as the lower body is being perfused by deoxygenated blood from the ductus arteriosus. PGE1 infusion will almost always reopen the ductus arteriosus and allow recovery of end-organ function prior to surgical repair (76). In the absence of data from randomized clinical trails, the method of nutritional support for these patients should be individualized and deserves comment. Those neonates with critical coarctation who present in shock with metabolic acidosis and laboratory evidence of end-organ dysfunction should be maintained on bowel rest prior to surgery, and nutritional support is provided with total parenteral nutrition. However, enteral feeds may be considered in the subset of infants who are diagnosed prior to the development of shock, started on PGE1, and subsequently have reasonable femoral pulses and diastolic blood pressure distal to the coarctation.
subclavian artery. A “posterior shelf” is present opposite the insertion site of the patent ductus arteriosus. Coarctation of the aorta may exist in isolation or can be associated with aortic arch hypoplasia, a VSD, or other complex intracardiac lesions. When present in isolation, coarctation of the aorta is “critical” when ductal dependent systemic blood flow exists, which represents only about 10% of cases. Neonates with isolated critical coarctation of the aorta also will present with shock if the diagnosis is not made before significant constriction of the ductus arteriosus occurs (Fig. 34-4). Prior to the development of shock, the diagnosis should be suspected by the detection of a brachial-femoral pulse discrepancy, which can be confirmed by measuring an arm-leg blood systolic pressure gradient. A systolic blood pressure gradient between the upper and lower extremities of greater than 10 mm Hg is clinically significant. In approximately 5% of infants with isolated coarctation of the aorta, the right subclavian artery will arise aberrantly from the descending thoracic aorta, distal to the coarctation. Thus it is important to palpate pulses and measure blood pressure in both upper extremities in all infants with suspected coarctation. Occasionally differential cyanosis will be present as the lower body is being perfused by deoxygenated blood from the ductus arteriosus. PGE1 infusion will almost always reopen the ductus arteriosus and allow recovery of end-organ function prior to surgical repair (76). In the absence of data from randomized clinical trails, the method of nutritional support for these patients should be individualized and deserves comment. Those neonates with critical coarctation who present in shock with metabolic acidosis and laboratory evidence of end-organ dysfunction should be maintained on bowel rest prior to surgery, and nutritional support is provided with total parenteral nutrition. However, enteral feeds may be considered in the subset of infants who are diagnosed prior to the development of shock, started on PGE1, and subsequently have reasonable femoral pulses and diastolic blood pressure distal to the coarctation.
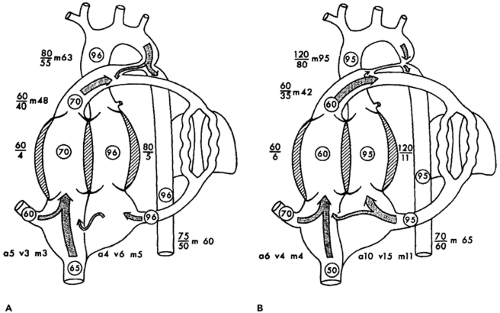 Figure 34-4 Schematic diagram of hemodynamic data and oxygen saturation levels in a neonate with critical coarctation of the aorta. A: Hemodynamic data and oxygen saturation levels prior to ductal closure. B: Hemodynamic data and oxygen saturation levels following ductal closure. Note the development of systemic hypertension and left atrial hypertension, the persistence of pulmonary hypertension, and the decrease in cardiac output as demonstrated by the lower oxygen saturation level in the inferior vena cava (a, atrial; v, ventricle; m, mean). Reprinted from Rudolph AM. Aortic arch obstruction. In: Congenital diseases of the heart: clinical-physiological considerations, 2nd ed. Armonk, NY: Futura, 2001:382-383, with permission. |
Hypoplastic Left Heart Syndrome
HLHS exists when there is severe mitral and aortic valve stenosis or atresia, a diminutive left ventricle, ascending aorta, and aortic arch, and often a juxtaductal coarctation of the aorta. Following birth there are two key communications that need to remain open to permit cardiopulmonary stability. A patent ductus arteriosus (PDA) is necessary to allow blood flow from the pulmonary artery to reach the systemic circulation, and shock will develop once the ductus arteriosus constricts. An adequate ASD is also necessary to allow egress of the pulmonary venous
blood flow from the left atrium to the right atrium. The 5% of infants with HLHS who have a severely restrictive or intact atrial septum will develop profound cyanosis, pulmonary edema, and shock almost immediately following delivery.
blood flow from the left atrium to the right atrium. The 5% of infants with HLHS who have a severely restrictive or intact atrial septum will develop profound cyanosis, pulmonary edema, and shock almost immediately following delivery.
Initially, most patients with HLHS (assuming an unrestrictive or mildly restrictive atrial communication) will be hemodynamically stable following the initiation of a PGE1 infusion. Over several days, however, as pulmonary vascular resistance falls, the ratio of pulmonary to systemic blood flow (Qp/Qs) will increase, leading to an increase in arterial oxygen saturation. Poor systemic circulation may manifest, and the neonate may develop right ventricular volume overload, and coronary and end-organ ischemia. The Qp/Qs may be calculated by the following equation which is derived from the Fick principle: Qp/Qs =(SaO2-SvO2)/(PvO2-PaO2), in which SaO2 =aortic saturation; SvO2 =mixed venous saturation (estimated by the superior vena cava oxygen saturation); PvO2 =pulmonary venous oxygen saturation (assumed to be > 95% if not directly measured); and PaO2 =pulmonary artery oxygen saturation (assumed equal to the aortic saturation). Most preoperative neonates will not have a catheter positioned in the superior vena cava to measure SvO2, and thus the Qp/Qs cannot be precisely calculated. Determination of an absolute value for Qp/Qs is of less importance than having an appreciation for the physiologic disturbance that develops as these neonates become progressively over circulated. Theoretical computer models of newborns with HLHS demonstrate that, beyond a certain point, slight increases in arterial oxygen saturation are associated with significant decreases in oxygen delivery, and that maximal oxygen delivery occurs at a Qp/Qs less than 1 (Fig. 34-5 and 34.6) (81,82). This model also demonstrates that a SvO2 less than 40%, or a SaO2-SvO2 difference of more than 40%, is likely associated with a severe derangement in systemic oxygen delivery, related to either a high Qp/Qs or low cardiac output (82).
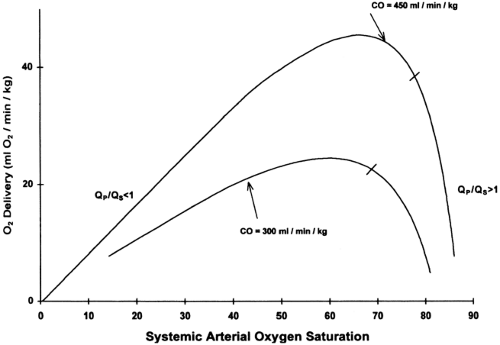 Figure 34-5 Systemic arterial oxygen saturation vs. systemic oxygen (O2) delivery in the theoretical newborn with hypoplastic left heart syndrome. A computer model was used to generate the curves by setting the cardiac output (CO) at 300 or 450 mL/kg/min and varying Qp/Qs from 0.2 to 10. The short line on each curve represents the point at which Qp/Qs = 1. As SaO2 increases, oxygen delivery increases and reaches a peak, and then decreases rapidly. Peak oxygen delivery occurs at a Qp/Qs 1. Reprinted from Barnea O, Santamore WP, Rossi A, et al. Estimation of oxygen delivery in newborns with a univentricular circulation. Circulation 1998;98:1407-13, with permission. |
In the typical neonate with HLHS, interventions that lower the pulmonary vascular resistance, such as supplemental oxygen and hyperventilation, may increase Qp/Qs and should be avoided. Patients with pulmonary over-circulation (high Qp/Qs) and evidence of poor end-organ perfusion (e.g., poor urine output, rising lactate levels, diminished peripheral pulses) may be treated with sub-ambient FiO2, obtained using supplemental inhaled nitrogen to increase pulmonary vascular resistance and thus restore the balance between systemic and pulmonary blood flow (83,84). The supplemental nitrogen may be delivered through the endotracheal tube or through a nasal cannula in neonates who are spontaneously breathing. Pulmonary vascular resistance may also be increased in such patients using supplemental inspired carbon dioxide (CO2) (85). In a prospective, randomized, crossover study, the impact of hypoxia (17% FiO2) vs. hypercarbia (2.7% FiCO2) on systemic oxygen delivery in preoperative neonates with HLHS was evaluated (86). All patients were receiving mechanical ventilation, deep sedation or anesthesia, and
neuromuscular blockade. The study found that both hypoxia and hypercarbia were effective in reducing Qp/Qs ratio (Fig. 34-7). Hypercarbia increased cerebral oxygen saturation, mean arterial blood pressure, oxygen delivery and, although not directly measured, cardiac output, whereas hypoxia did not change these variables, suggesting that hypercarbia may be the preferred therapy (Fig. 34-8) (86,87). However, administration of supplemental CO2 requires the concurrent use of sedation and paralysis, thus limiting its applicability for most neonates with HLHS prior to surgery.
neuromuscular blockade. The study found that both hypoxia and hypercarbia were effective in reducing Qp/Qs ratio (Fig. 34-7). Hypercarbia increased cerebral oxygen saturation, mean arterial blood pressure, oxygen delivery and, although not directly measured, cardiac output, whereas hypoxia did not change these variables, suggesting that hypercarbia may be the preferred therapy (Fig. 34-8) (86,87). However, administration of supplemental CO2 requires the concurrent use of sedation and paralysis, thus limiting its applicability for most neonates with HLHS prior to surgery.
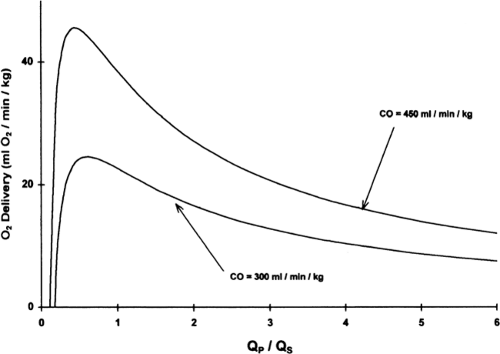 Figure 34-6 Systemic oxygen (O2) delivery vs. Qp/Qs in the theoretical newborn with hypoplastic left heart syndrome. A computer model was used to generate the curves by setting the cardiac output (CO) at 300 or 450 mL/kg/min. Note that increasing cardiac output can increase oxygen delivery, and that oxygen delivery decreases significantly once Qp/Qs exceeds 1. Reprinted from Barnea O, Santamore WP, Rossi A, et al. Estimation of oxygen delivery in newborns with a univentricular circulation. Circulation 1998;98:1407-1413, with permission. |
Neonates with HLHS and myocardial dysfunction should receive inotropic support to improve cardiac output and systemic oxygen delivery. However, care should be taken to avoid inotropic agents that increase systemic vascular resistance because this will favor pulmonary blood flow over systemic blood flow. Low-dose dopamine or milrinone are good inotropic agents for neonates with HLHS and myocardial dysfunction.
The neonate with HLHS and a severely restrictive or intact atrial septum will present with profound cyanosis and shock immediately after birth as a result of obstruction of blood egress from the left atrium. The short- and long-term outlook for such infants has historically been poor, regardless of whether they undergo the Norwood operation or are listed for heart transplantation, due in large part to the in utero development of pulmonary venous hypertension and lymphatic abnormalities that persist following birth (88,89,90). The immediate management of these critically ill neonates involves prompt intervention to decompress the left atrium. Emergent surgical intervention, involving either open atrial septectomy or a Norwood procedure, has usually resulted in early death. A Brockenbrough atrial septoplasty (transcatheter, trans-septal needle puncture followed by serial balloon dilation of the new ASD and possibly stent placement) will serve to decompress the left atrium, increase pulmonary venous return and pulmonary blood flow, and alleviate cyanosis (35). Following this procedure, the patient may be medically managed for a few days with a PGE1 infusion and diuretics to allow pulmonary vascular resistance to fall and pulmonary edema to improve prior to the Norwood operation. This multidisciplinary approach to the neonate with HLHS and severely restrictive or intact atrial septum has resulted in a contemporary 53% survival rate following the Norwood operation, which is better than results obtained with other management strategies for this lesion, but remains poor when compared with the outcome of neonates without atrial obstruction who undergo the Norwood operation (91). Theoretically, alleviation of the left atrial hypertension in utero may improve outcome in patients with HLHS and a severely restrictive or intact atrial septum. Fetal intervention to dilate the atrial septum using percutaneous, transcatheter needle puncture of the atrial septum followed by balloon dilation appears to be feasible and is associated with minimal maternal risk, but technical improvements and additional experience will be required before this procedure is widely adopted (92).
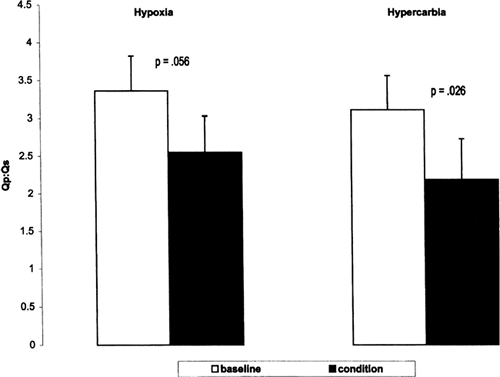 Figure 34-7 Difference in Qp/Qs between condition (either hypoxia or hypercarbia) and baseline (mean ± SEM) in preoperative neonates with hypoplastic left heart syndrome. Reprinted from Tabbutt S, Ramamoorthy C, Montenegro LM, et al. Impact of inspired gas mixtures on preoperative infants with hypoplastic left heart syndrome during controlled ventilation. Circulation 2001; 104:I159-I164, with permission. |
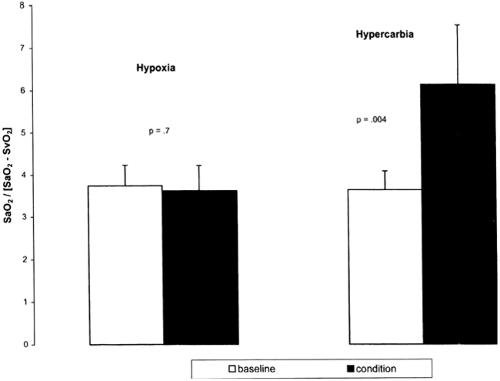 Figure 34-8 Difference in oxygen delivery between condition (either hypoxia or hypercarbia) and baseline (mean ± SEM) in preoperative neonates with hypoplastic left heart syndrome. Oxygen delivery was calculated as SaO2 Ö (SaO2-SvO2), in which SaO2 and SvO2 were directly measured. Reprinted from Tabbutt S, Ramamoorthy C, Montenegro LM, et al. Impact of inspired gas mixtures on preoperative infants with hypoplastic left heart syndrome during controlled ventilation. Circulation 2001;104:I159-I164, with permission. |
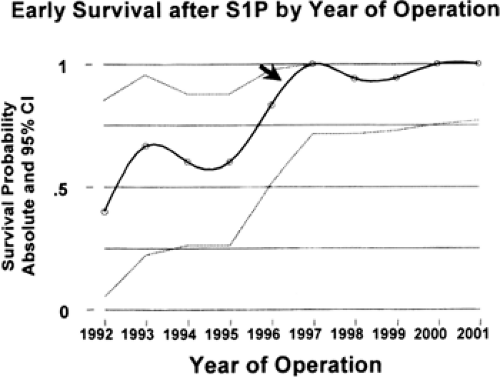 Figure 34-9 Hospital survival following stage 1 palliation in patients with hypoplastic left heart syndrome by year at a single center. Hospital survival improved coincident with the application of new treatment strategies beginning in July of 1996 (arrow). S1P, stage 1 palliation (Norwood operation). Reprinted from Tweddell JS, Hoffman GM, Mussatto KA, et al. Improved survival of patients undergoing palliation of hypoplastic left heart syndrome: lessons learned from 115 consecutive patients. Circulation 2002;106: I82-I89, with permission. |
Initial reports suggested that severe tricuspid regurgitation was a significant risk factor for infants with HLHS (93,94). However, in the preoperative infant with HLHS, the severity of tricuspid regurgitation may be in part related to a high Qp/Qs, with right ventricular volume overload and stretching of the tricuspid valve annulus. Following the Norwood operation, the Qp/Qs may decrease as a result of the fixed resistance to pulmonary blood flow provided by the shunt, and subsequently the degree of tricuspid regurgitation may diminish. Thus, clinical judgment and an appreciation of tricuspid valve morphology are required in such cases.
 Figure 34-10 The three types of interrupted aortic arch. In type A, the interruption is at the aortic isthmus between the left subclavian artery and the ductus. In type B, the interruption is at the distal aortic arch between the left carotid and left subclavian arteries. In type C, the interruption is at the proximal aortic arch between the innominate and left carotid arteries. Type B is the most common form of this lesion. Type C is rare. Reprinted from Chang AC, Starnes VA. Interrupted aortic arch. In: Chang AC, Hanley FL, Wernovsky G, et al., eds. Pediatric cardiac intensive care. Baltimore: Williams and Wilkins, 1998:243-247, with permission. |
Several centers have recently reported overall survival rates of more than or equal to 90% following initial surgical palliation for HLHS (Fig. 34-9) (4,95). Contemporary risk factors contributing to higher mortality following the Norwood operation include prematurity, the presence of multiple congenital anomalies, and an intact atrial septum (48,88). Although palliative care is still offered at some centers for neonates with HLHS, fewer clinicians and parents are opting for no intervention given the improving surgical results (96). Some centers offer heart transplantation as a primary option for HLHS (97). Problems with this strategy include the high morbidity and mortality incurred while waiting for a heart to become available from a very limited donor pool, suboptimal neurologic outcome in many patients, and complications inherent to heart transplant (98,99,100). For these reasons, many pediatric cardiology centers recommend the Norwood operation as the initial surgical intervention in neonates with HLHS, and heart transplantation is typically reserved for the small subset that develop subsequent irreversible myocardial failure.
Interrupted Aortic Arch
An interrupted aortic arch (IAA) is most commonly associated with a VSD, but also is seen with a variety of other congenital cardiac malformations, including truncus arteriosus and TGA (101). If a VSD is not present in a patient with an IAA, an aorto-pulmonary window is likely to be found. IAA is classified by the location of the arch interruption in relation to the bracheocephalic vessels (Fig. 34-10) (102). IAA type B is the most common subtype (>50% of cases), and type C is rare. Chromosome 22q11 deletion is present in approximately 60% of neonates with IAA type B (41). The infant with an unrecognized IAA will present with shock, similar to a critical coarctation, upon closure of the ductus arteriosus. Infants with IAA are dependent upon ductal patency to maintain blood flow to the lower body, and
preoperative management strategies to balance the systemic and pulmonary circulations mimic those used in infants with HLHS. Early surgical intervention is warranted.
preoperative management strategies to balance the systemic and pulmonary circulations mimic those used in infants with HLHS. Early surgical intervention is warranted.
Mixing Lesions
Transposition of the Great Arteries
In d-TGA, the aorta arises from the anatomic right ventricle and the pulmonary artery arises from the anatomic left ventricle. Approximately 40% of neonates with TGA have an associated VSD, which occasionally is a malalignment type defect. If anterior malalignment exists, there may be associated right ventricular outflow tract obstruction, aortic valvar stenosis, coarctation of the aorta or rarely an IAA. A posterior malalignment VSD is associated with left ventricular outflow tract obstruction, pulmonary stenosis or atresia. Coronary artery branching abnormalities are present in approximately 30% of cases.
In this unique parallel circulation, deoxygenated systemic venous blood returns to the right heart and is pumped back to the systemic arterial circulation, and oxygenated pulmonary venous blood passes through the left heart and is pumped back to the lungs. Unless there is adequate mixing between these parallel circulations, severe cyanosis, metabolic acidosis and death will occur. Such intercirculatory mixing represents effective pulmonary and systemic blood flows, and may take place at the atrial, ventricular, or great artery level (Fig. 34-11). Infants with TGA and an intact ventricular septum typically have a patent foramen ovale (PFO) or ASD that allows some mixing at the atrial level. However, a PGE1 infusion is usually necessary to open or maintain patency of the ductus arteriosus, which will increase effective pulmonary blood flow, provided that the pulmonary vascular resistance is lower than the systemic vascular resistance and that there is an adequate atrial communication.
Occasionally infants with TGA may develop severe cyanosis and shock following the initiation of PGE1 infusion, as a result of increasing blood volume returning to the left atrium and functional closure of the foramen ovale. If this occurs, or if the PFO is restrictive, as assessed by echocardiography, and excessive cyanosis is present, an emergent balloon atrial septostomy should be performed to enlarge the atrial communication (Fig. 34-12, see also Color Plate) (34). The balloon atrial septostomy may be performed at the bedside using echocardiographic guidance, or in the cardiac catheterization laboratory (103). Excessive cyanosis may persist despite a technically successful balloon atrial septostomy. High pulmonary vascular resistance may limit effective pulmonary blood flow, and measures should be taken to lower pulmonary vascular resistance. Because the majority of systemic blood flow comes from the systemic venous circulation in TGA, neonates who remain excessively cyanotic following a balloon atrial septostomy will also improve following maneuvers that will increase the mixed venous oxygen saturation. These include interventions to decrease oxygen consumption, such as sedation and paralysis, and improve oxygen delivery, such as correcting anemia and administering inotropic agents. To summarize, infants with TGA and intact ventricular septum who remain excessively cyanotic despite the initiation of PGE1 require a balloon atrial septostomy, and if necessary, interventions to lower pulmonary vascular resistance and increase systemic venous oxygen saturation.
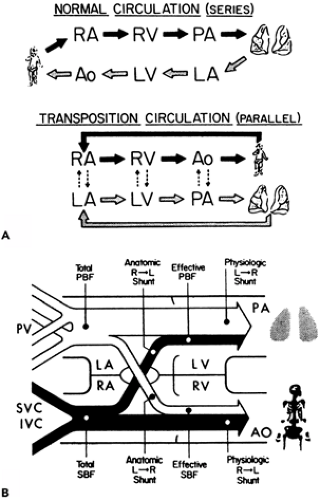 Figure 34-11 The circulation in transposition of the great arteries. A: The systemic and pulmonary circulations are in series in the normal circulation, whereas they are in parallel in TGA. Solid arrows, relatively unoxygenated blood; stippled arrows, oxygenated blood; dashed arrows, intercirculatory shunts. B: Circulation schema demonstrating flows and shunts in infants with TGA/intact ventricular septum. Note that the anatomic left to right shunt constitutes the effective SBF, and the anatomic right-to-left shunt constitutes the effective PBF. Ao, aorta; IVC, inferior vena cava; LA, left atrium; LV, left ventricle; L → R, left to right; RA, right atrium; RV, right ventricle; R → L, right to left; PA, pulmonary artery; PBF, pulmonary blood flow; PV, pulmonary veins; SBF, systemic blood flow; SVC, superior vena cava. Reprinted from Paul MH, Wernovsky G. Transposition of the great arteries. In: Emmanouilides GC, Riemenschneider TA, Allen HD, et al, eds. Moss and Adams’ heart disease in infants, children and Adolescents, including the fetus and young adult. Baltimore: Williams & Wilkins, 1995:1154-1225, with permission. |
Many clinicians recommend that a semi-elective balloon atrial septostomy be performed in nearly all patients with TGA and intact ventricular septum, even those without
excessive cyanosis. Once the atrial septum is enlarged, the PGE1 can usually be safely discontinued, thus avoiding complications related to that medication and a large ductus arteriosus, such as apnea and NEC. Furthermore, the left atrium is decompressed following a balloon atrial septostomy, which may allow the pulmonary vascular resistance to fall prior to surgery. Finally, the infant may be transferred from the ICU to the general ward to initiate feedings and await surgery. The major risks associated with the performance of a balloon atrial septostomy include myocardial perforation or avulsion of the inferior vena cava from the right atrium during pullback of the balloon, both of which rarely occur. Following initial stabilization of the neonate with TGA and intact ventricular septum, an early arterial switch operation is required before the left ventricle becomes deconditioned.
excessive cyanosis. Once the atrial septum is enlarged, the PGE1 can usually be safely discontinued, thus avoiding complications related to that medication and a large ductus arteriosus, such as apnea and NEC. Furthermore, the left atrium is decompressed following a balloon atrial septostomy, which may allow the pulmonary vascular resistance to fall prior to surgery. Finally, the infant may be transferred from the ICU to the general ward to initiate feedings and await surgery. The major risks associated with the performance of a balloon atrial septostomy include myocardial perforation or avulsion of the inferior vena cava from the right atrium during pullback of the balloon, both of which rarely occur. Following initial stabilization of the neonate with TGA and intact ventricular septum, an early arterial switch operation is required before the left ventricle becomes deconditioned.
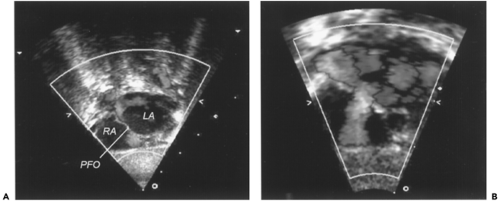 Figure 34-12 Restrictive atrial septum in a newborn with transposition of the great arteries. A. Two-dimensional echocardiogram with color Doppler from the subcostal window demonstrating a tiny patent foramen ovale with left to right flow across the atrial septum. B. Following successful balloon atrial septostomy, a wide communication now exists between the left and right atria. LA, left atrium; PFO, patent foramen ovale; RA, right atrium. (See color plate) |
Infants with TGA and moderate to large VSD generally are well oxygenated and do not require a PGE1 infusion provided that the ventricular outflow tracts are unobstructed. Early congestive heart failure typically develops, and surgical repair is indicated within the first few weeks of life. A malalignment-type VSD may be associated with clinically significant left or right ventricular outflow tract obstruction. Patients with sub-aortic obstruction may have an associated coarctation of the aorta or an IAA. Such an infant may present with the very unique finding of reverse differential cyanosis, with low oxygen saturation in the right arm and high oxygen saturation in the leg. A PGE1 infusion is needed, followed by early surgical intervention. Infants with d-TGA and VSD who are referred for surgery after the first few months of life (typically from underdeveloped nations) are at risk for having developed pulmonary vascular obstructive disease. These patients may require a cardiac catheterization to evaluate their pulmonary vascular resistance prior to surgery.
Tricuspid Atresia
Tricuspid atresia is present when there is agenesis of the tricuspid valve and an absent communication between the right atrium and right ventricle. The presentation of infants with this lesion is variable and depends primarily upon the presence and size of a VSD, whether the great arteries are normally related or transposed, and the degree of ventricular outflow tract obstruction (Table 34-5) (104,105). These factors determine which of these neonates will be dependent on a PGE1 infusion. All of the systemic venous blood must pass through an obligate atrial communication to the left atrium, in which it mixes with the pulmonary venous blood. In the presence of normally looped ventricles and normally related great arteries, the left ventricle then ejects blood to the aorta, and through the VSD, if present, to the pulmonary artery. If the great arteries are transposed, the left ventricle pumps blood to the pulmonary artery (unless pulmonary atresia is present), and through the VSD to the aorta. Although all infants with tricuspid atresia eventually require a Fontan palliation, the type of initial operation depends upon the extent of cyanosis or systemic outflow obstruction. For example, infants with tricuspid atresia, normally looped ventricles and normally related great vessels, a large VSD and no or minimal right ventricular outflow tract obstruction (type I-C) will be well oxygenated and develop early heart failure. In the neonatal period, they require placement of a pulmonary artery band or transection of the pulmonary artery and placement of a systemic to pulmonary artery shunt. Those with a moderate size VSD and a moderate degree of right ventricular outflow tract obstruction (type I-B) will have a balanced circulation with an acceptable amount of cyanosis. Such infants may be discharged home with the expectation of a bi-directional
Glenn or hemi-Fontan operation within the first six months of life. Infants with tricuspid atresia and a small or absent VSD (type I-A or I-B) will have significant cyanosis and thus be dependent on a PGE1 infusion until a systemic to pulmonary artery shunt is performed. Infants with tricuspid atresia and transposed great arteries, a small or absent VSD (type II-A or II-B) will develop shock unless ductal patency is maintained. These infants require a Damus-Kaye-Stansel operation (aortopulmonary anastomosis) or a modified Norwood operation with a systemic to pulmonary artery shunt.
Glenn or hemi-Fontan operation within the first six months of life. Infants with tricuspid atresia and a small or absent VSD (type I-A or I-B) will have significant cyanosis and thus be dependent on a PGE1 infusion until a systemic to pulmonary artery shunt is performed. Infants with tricuspid atresia and transposed great arteries, a small or absent VSD (type II-A or II-B) will develop shock unless ductal patency is maintained. These infants require a Damus-Kaye-Stansel operation (aortopulmonary anastomosis) or a modified Norwood operation with a systemic to pulmonary artery shunt.
TABLE 34-5 CLASSIFICATION SYSTEM FOR TRICUSPID ATRESIA | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Total Anomalous Pulmonary Venous Return
Total anomalous pulmonary venous return (TAPVR) is a congenital heart defect in which all four pulmonary veins drain to a systemic vein or the right atrium. TAPVR results from a failure of the common pulmonary vein to fuse with the posterior surface of the left atrium early in fetal life. The pulmonary veins then decompress via the coronary sinus or primitive venous structures, which eventually lead to the right atrium, in which mixing occurs with the systemic venous blood. An anatomic classification system divides TAPVR into supracardiac, cardiac, infracardiac, and mixed, depending upon which primitive venous pathways are employed. Supracardiac TAPVR exists when all of the pulmonary veins come to a confluence behind the left atrium and drain via a vertical vein to the innominate vein or superior vena cava. Occasionally the vertical vein is obstructed as it passes between the left mainstem bronchus and left pulmonary artery. Cardiac TAPVR exists when the pulmonary veins drain to the coronary sinus or right atrium. Obstruction is uncommon in cardiac TAPVR. In infracardiac TAPVR, the pulmonary venous confluence drains inferiorly through the diaphragm and into the portal or hepatic veins, in which obstruction is common. Patients with mixed TAPVR have drainage to more than one site. For example, the left sided pulmonary veins may drain through a vertical vein to the innominate vein, although the right pulmonary veins drain to the right atrium. An obligatory intracardiac (usually atrial) communication exists in neonates with TAPVR to allow some oxygenated blood to reach the left heart and systemic arterial circulation. TAPVR may be associated with significant intracardiac disease as is seen in patients with heterotaxy syndrome, or may exist as an isolated lesion.
The presentation of isolated TAPVR is primarily based upon the presence and degree of obstruction between the pulmonary veins and the right heart. Most patients with infracardiac TAPVR and some with supracardiac TAPVR will have obstructed pulmonary venous pathways that result in pulmonary edema, pulmonary hypertension, cyanosis, and significant respiratory distress soon after birth. The clinical presentation and CXR may mimic those seen with neonatal pneumonia or respiratory distress syndrome, leading to delays in diagnosis. If a venous blood gas is drawn from an umbilical venous catheter positioned just above the liver in a cyanotic newborn, and the oxygen saturation level is in the high 90s, the diagnosis of infracardiac TAPVR should be suspected, as the sample is reflective of pulmonary venous return. The umbilical venous catheter should be removed so that it does not further obstruct pulmonary venous flow and cause clinical decompensation. Stabilization for the neonate with obstructed TAPVR involves mechanical ventilation, sedation and urgent surgical intervention (106). Hypoxemia may be abated somewhat by interventions that increase mixed venous oxygen saturation, including pharmacologic paralysis, correction of anemia, and inotropic support. Although neonates with obstructed TAPVR may be quite cyanotic, measures to lower pulmonary vascular resistance, including the use of hyperventilation and nitric oxide (NO), are usually not beneficial. The use of PGE1 infusion in such patients is controversial, but generally is discouraged. Some physicians feel that PGE1 may maintain patency of the ductus venosus in infants with infracardiac obstructed TAPVR, thus allowing for better drainage back to the right heart. Other clinicians believe that maintaining patency of the ductus arteriosus may allow left to right shunting at the arterial level (if the pulmonary vascular resistance is less than the systemic vascular resistance) and thus exacerbate pulmonary edema.
Infants with unobstructed isolated TAPVR and an adequate atrial communication will be mildly hypoxemic without overt clinical cyanosis at birth but develop congestive heart failure (CHF) within a few weeks to months. Surgical repair is performed within the first few days to weeks of life.
Truncus Arteriosus
Truncus arteriosus is present when a single (common) arterial trunk gives rise to the aorta, at least one coronary artery, and at least one pulmonary artery. A single semilunar (“truncal”) valve is always present, and an unrestrictive VSD is almost always present. About one-third of patients with truncus arteriosus have a 22q11.2 microdeletion (DiGeorge syndrome) that can be diagnosed with florescent in-situ hybridization (FISH) (107). Several classification systems exist that may be used to describe the origins of the pulmonary arteries from the arterial trunk and the presence or absence of an IAA (108). Newborns with truncus arteriosus who present with mild cyanosis do not require a PGE1 infusion unless an IAA (about 10% of cases) or ductal origin of one of the pulmonary arteries is present.
Infants with truncus arteriosus usually develop too much pulmonary blood flow and congestive heart failure within a few weeks after birth as pulmonary vascular resistance falls and systemic vascular resistance increases, unless stenosis is present at the origin of the pulmonary arteries from the aorta. Supplemental oxygen should be avoided to minimize the risk of decreasing pulmonary vascular resistance. Congestive heart failure is exacerbated in the presence of significant truncal valve insufficiency, which occurs in 50% of cases, or significant truncal valve stenosis (less common). For this reason, and to prevent the development of pulmonary vascular obstructive disease before surgery and minimize the incidence of pulmonary hypertension crises postoperatively, complete surgical repair is generally performed prior to discharge from the nursery (8,109,110).
Left to Right Shunts
Atrial Septal Defect
ASDs may be present in isolation or with nearly any other type of congenital heart defect. ASDs are categorized by location and include secundum, ostium primum and sinus venosus types. The most common type is the secundum ASD, which exists when there is a deficiency in the septum primum in the central portion of the atrial septum. Although secundum ASDs may become smaller and close spontaneously, particularly those that are less than 5 mm in diameter, the ostium primum and sinus venosus defects will always require surgical intervention. The ostium primum defects are located in the inferior region of the atrial septum, and are almost always associated with an additional endocardial cushion abnormality. The sinus venosus defects usually occur at the junction between the superior vena cava and the right atrium, and are almost always associated with partial anomalous pulmonary venous return.
Isolated ASDs only rarely cause symptoms in infants, as significant left to right shunting does not occur until right ventricular compliance falls below left ventricular compliance, and this often takes years to occur. Likewise it is uncommon for patients with large ASDs to require oral medications (digoxin or diuretics). However, clinical judgment regarding the medical management and timing of surgical intervention is required in the neonate with a large ASD and additional medical problems (e.g., chronic lung disease) that complicate the clinical picture. The primary indication for surgery in infancy is the presence of symptoms secondary to left to right shunting. Toddlers require intervention if they have an ostium primum or sinus venosus ASD, or a secundum ASD with evidence for volume overload of the right ventricle. Intervention is indicated in early childhood in asymptomatic patients with significant left to right atrial shunts to prevent late complications including pulmonary vascular obstructive disease, thromboembolic events, arrhythmias, and right heart failure. A variety of devices exist that are used to close secundum ASDs in the catheterization laboratory, but the majority of clinical experience thus far exists with older children (111).
Ventricular Septal Defect
VSDs may be found in a variety of complex congenital heart lesions, or may exist in isolation. VSDs are categorized by location and size. VSDs may be located in the perimembranous, muscular, inlet or conal (outlet, subarterial, supracristal) regions of the ventricular septum, and there may be more than one VSD present. Malalignment of the conal septum may exist, resulting in variable degrees of right or left ventricular outflow tract obstruction. Conal VSDs often develop involvement of the aortic valve over many years (112). The size may be determined by an estimation of the diameter of the VSD in relation to the aortic annulus, and by degree of restriction to blood flow as estimated by Doppler echocardiogram. Perimembranous and muscular VSDs may become smaller or close with time, particularly if the initial diameter of the VSD is less than 5 mm (113). In patients with isolated moderate to large VSDs, congestive heart failure develops over time once an imbalance exists between systemic and pulmonary vascular resistance and a significant left to right shunt develops. Neonates with large VSDs will typically develop pulmonary overcirculation, left ventricular volume overload, and symptoms of congestive heart failure within the first several weeks to months of life as pulmonary vascular resistance falls. Any associated left ventricular outflow tract obstruction, such as coarctation of the aorta, will exacerbate this problem. Premature infants with left to right shunting at the ventricular or great artery level may develop congestive heart failure sooner as a result of decreased musculature in the pulmonary arterioles that allows for a precipitous drop in pulmonary vascular resistance.
A trial of medical management is usually indicated in infants with moderate to large VSDs. Provision of adequate nutrition and administration of medications including
loop diuretics and sometimes digoxin and/or angiotensin converting enzyme inhibitors (ACE inhibitors) can often allow surgery to be deferred for a few months (114). Supplemental oxygen and hyperventilation should be avoided, as they will cause pulmonary vascular resistance to fall, and Qp/Qs to rise. Infants who continue to have failure to thrive despite these measures are referred for surgical repair, or in selected cases, transcatheter intervention (115).
loop diuretics and sometimes digoxin and/or angiotensin converting enzyme inhibitors (ACE inhibitors) can often allow surgery to be deferred for a few months (114). Supplemental oxygen and hyperventilation should be avoided, as they will cause pulmonary vascular resistance to fall, and Qp/Qs to rise. Infants who continue to have failure to thrive despite these measures are referred for surgical repair, or in selected cases, transcatheter intervention (115).
Atrioventricular Canal Defect
The presence or absence of a common atrioventricular valve oriface and the relative size of the physiologic VSD characterizes the different types of atrioventricular septal defects, also known as endocardial cushion defects. In patients with an incomplete atrioventricular canal, an ostium primum ASD exists along with a cleft in the mitral valve. There is no physiologic VSD present. A transitional atrioventricular canal defect has an ostium primum ASD, dense attachments of the atrioventricular valve leaflets to the crest of the ventricular septum such that two functionally separate atrioventricular valves exist, and a small physiologic VSD. A complete atrioventricular canal defect has a primum ASD, a common atrioventricular valve and a large inlet VSD. Associate anomalies include a left superior vena cava to the coronary sinus, additional VSDs, and aortic arch obstruction. Uncommonly, right ventricular outflow tract obstruction is seen, and such patients have features of tetralogy of Fallot and an atrioventricular canal (116). In most cases the ventricles are balanced, such that both are of adequate size to support the full cardiac output following surgery. Occasionally one of the ventricles is hypoplastic (“unbalanced”), precluding a two ventricular repair. Such patients may be considered for a one and a half ventricular repair, the single ventricle pathway, or heart transplantation.
Infants with complete atrioventricular canal defects typically develop congestive heart failure within the first few weeks to months of life. The congestive heart failure will be exacerbated if significant atrioventricular valve regurgitation or aortic arch hypoplasia is present. There is an increased incidence of Down syndrome (trisomy 21) in patients with endocardial cushion defects, and such patients are predisposed to the early development of pulmonary vascular obstructive disease. A trial of medical management is indicated in symptomatic infants, as discussed above for patients with large VSDs. Surgical repair is required within the first three to six months of life to eliminate the symptoms of congestive heart failure, prevent the development of pulmonary vascular obstructive disease and minimize the incidence of postoperative pulmonary hypertensive crises. Surgical repair is not commonly performed in the newborn period as a result of the difficulty with suturing the paper-thin atrioventricular valve leaflets.
Infants with incomplete or transitional atrioventricular canal defects often follow the clinical course of patients with isolated ASDs, and these lesions are typically repaired sometime between 6 months and 4 years of age.
Patent Ductus Arteriosus
The ductus arteriosus normally is functionally closed within the first hours to days of life. PDAs may exist in isolation or with many other types of congenital heart disease. An isolated, large PDA will cause symptoms of congestive heart failure once the pulmonary vascular resistance falls, allowing a left to right shunt from the aorta to the pulmonary artery. Left atrial and left ventricular volume overload are present and pulmonary edema may occur. Diastolic runoff exists leading to a “steal” from systemic blood flow, which may contribute to mesenteric ischemia and the development of NEC. PDAs are more common and cause more symptoms in premature infants. Small PDAs typically do not cause symptoms but are a long-term risk for bacterial endocarditis. Symptoms of congestive heart failure may be treated with diuretics, digoxin and the provision of adequate nutrition. Approximately two-thirds of PDAs in premature infants will close following a course of indomethacin or ibuprofen (117). Surgical ligation is indicated for PDAs that do not respond to medical therapy in premature infants, and for PDAs in any symptomatic term infant. Older children with a continuous murmur but no symptoms should have the PDA closed surgically or in the cardiac catheterization laboratory to minimize the risk of endocarditis.
Aortopulmonary Window
An aortopulmonary window is an uncommon, usually large communication between the ascending aorta and pulmonary artery in the presence of two semilunar valves. This lesion usually exists in isolation, but may be associated with IAA, VSD, or other congenital heart defects (118,119). Infants with large aortopulmonary windows will usually develop congestive heart failure at several weeks of life. Prompt surgical repair is indicated in most cases to eliminate the symptoms of congestive heart failure, prevent the development of pulmonary vascular obstructive disease and minimize the incidence of postoperative pulmonary hypertension.
Cardiomyopathy and Heart Transplantation
Cardiomyopathy
A multidisciplinary team, lead by the intensivist, a cardiologist with expertise in heart failure and transplantation, and a congenital heart surgeon, is required to answer several key questions for each infant who presents with a severe cardiomyopathy: What is the primary cause for the cardiomyopathy? Is it treatable or reversible? What level of medical or mechanical support is required? Is the infant a candidate for heart transplantation?
Infants may develop a severe cardiomyopathy from a wide variety of causes, including infection, structural heart disease, chronic arrhythmia, metabolic disorders, and primary myopathies. The broad differential diagnosis for infantile
cardiomyopathies is covered in detail in Chapter 33 and will not be repeated here. However, several conditions that may present with severe cardiomyopathy, are difficult to diagnose and are sometimes erroneously referred for heart transplantation, yet are readily treatable deserve comment. Infants born with anomalous left coronary artery from the pulmonary artery (ALCAPA) develop coronary ischemia when the pulmonary artery pressures fall after birth. These patients typically present in the first few weeks to months of life with severe left ventricular dysfunction, mitral regurgitation (as a result of papillary muscle ischemia), and pulmonary edema. The diagnosis may be suspected by the presence of characteristic findings on the electrocardiogram, and confirmed by echocardiography with careful examination of the coronary anatomy and blood flow pattern (120,121). Following reimplantation of the left coronary artery in the aorta, the vast majority of infants with ALCAPA recover left ventricular and mitral valve function (122). Infants with incessant arrhythmias, such as automatic atrial tachycardia, may present with severe cardiomyopathy. The initial rhythm may be thought to be sinus tachycardia secondary to the cardiomyopathy, but the correct diagnosis may be made by careful examination of the P wave morphology on the electrocardiogram. Myocardial function will recover once the arrhythmia is controlled with medications or ablation. Severe viral myocarditis is another condition that may be difficult to differentiate from a primary cardiomyopathy. Myocardial biopsy may provide clarity but is not without risk in sick infants. Many infants with viral myocarditis will recover ventricular function over time with supportive care (123).
cardiomyopathies is covered in detail in Chapter 33 and will not be repeated here. However, several conditions that may present with severe cardiomyopathy, are difficult to diagnose and are sometimes erroneously referred for heart transplantation, yet are readily treatable deserve comment. Infants born with anomalous left coronary artery from the pulmonary artery (ALCAPA) develop coronary ischemia when the pulmonary artery pressures fall after birth. These patients typically present in the first few weeks to months of life with severe left ventricular dysfunction, mitral regurgitation (as a result of papillary muscle ischemia), and pulmonary edema. The diagnosis may be suspected by the presence of characteristic findings on the electrocardiogram, and confirmed by echocardiography with careful examination of the coronary anatomy and blood flow pattern (120,121). Following reimplantation of the left coronary artery in the aorta, the vast majority of infants with ALCAPA recover left ventricular and mitral valve function (122). Infants with incessant arrhythmias, such as automatic atrial tachycardia, may present with severe cardiomyopathy. The initial rhythm may be thought to be sinus tachycardia secondary to the cardiomyopathy, but the correct diagnosis may be made by careful examination of the P wave morphology on the electrocardiogram. Myocardial function will recover once the arrhythmia is controlled with medications or ablation. Severe viral myocarditis is another condition that may be difficult to differentiate from a primary cardiomyopathy. Myocardial biopsy may provide clarity but is not without risk in sick infants. Many infants with viral myocarditis will recover ventricular function over time with supportive care (123).
Infants with cardiomyopathy may present with congestive heart failure or shock. Many of the principles for supporting cardiac output and monitoring of patients with severe cardiomyopathy are discussed below in the “postoperative care” section, and thus will not be reiterated. Mechanical support, typically with venoarterial extracorporeal membrane oxygenation (ECMO) in infants, may be used as a bridge to recovery or transplantation and is discussed further below. Indications for mechanical support in an infant with severe cardiomyopathy include worsening end-organ function and metabolic acidosis despite maximal medical support.
TABLE 34-6 CONGENITAL HEART DEFECTS THAT MAY WARRANT PRIMARY HEART TRANSPLANTATION | ||
|---|---|---|
|
Heart Transplantation
Heart transplantation is a widely accepted option for many infants with severe congenital heart disease and irreversible cardiomyopathies. Approximately 100 infant heart transplants are reported annually to the registry of the International Society for Heart and Lung Transplantation. Seventy-five percent of these transplants are for congenital heart disease, and the majority of the remainder are for cardiomyopathies (124). Heart transplantation is considered a primary option in some institutions for infants with complex congenital heart disease if surgical intervention is believed to result in an unacceptably high mortality rate (Table 34-6). Transplantation is also an option if uncorrectable anatomical issues and/or irreversible myocardial dysfunction are present following initial surgical intervention. The donor pool is very limited, and as a result as many as 25% of infants will die while waiting for a heart to become available. If more institutions adopted a policy of primary transplantation for the lesions listed in Table 34-6, particularly HLHS, the mortality while waiting for a donor heart to become available would certainly increase. Recent experience with ABO incompatible transplantation has shortened the wait times at a few centers (125). Relative contraindications to heart transplantation include high, fixed pulmonary vascular resistance (>6-8 Woods units per m2), recent or recurrent malignancy, serious infection, significant systemic disease, other organ system disease (e.g., hepatic, renal, or neurological), and chromosomal, metabolic or genetic abnormalities with a poor long-term prognosis.
Although the 1-year survival following heart transplantation in infancy is lower than that reported for older children, the long-term survival appears to be better, perhaps related to immune tolerance or better compliance with medications (124). The 10-year survival following infant heart transplantation is approximately 60%. Infants who have a successful heart transplant face potential long-term complications related to immunosuppression (i.e., renal insufficiency, infection, diabetes mellitus, and malignancy), transplant coronary artery disease, and rejection (124). In depth conversations about these issues
with parents are always required before an infant is listed for transplantation.
with parents are always required before an infant is listed for transplantation.
Comprehensive supportive care is required to maintain patients who are awaiting transplantation in the ICU. An appropriate level of cardiopulmonary support is required to maintain end-organ function. This often entails the use of inotropic agents, vasodilators, and diuretics. Mechanical support, usually ECMO in infants, is occasionally required (126).
Mechanical ventilation may be beneficial to minimize oxygen consumption, and positive end-expiratory pressure will reduce wall stress and thus provide afterload reduction for a failing systemic ventricle. Close attention must also be given to the prevention of infection and the provision of adequate nutrition. Anticoagulation may be indicated for infants with severe myocardial dysfunction to minimize the chance of luminal clot formation.
Management of the infant with HLHS awaiting transplantation deserves comment. Ductal patency is maintained with a low dose PGE1 infusion, and occasionally these infants can be discharged to home while awaiting transplantation (127). Infants with HLHS awaiting heart transplantation tend to develop pulmonary overcirculation as pulmonary vascular resistance falls over time, which further volume overloads the right ventricle and may exacerbate tricuspid regurgitation. The extended use of subambiant FiO2, achieved using supplemental inspired nitrogen, to increase pulmonary vascular resistance and limit pulmonary blood flow has been reported in this patient population (84,127). Limitation of pulmonary blood flow by banding of the pulmonary artery, either surgically or using innovative transcatheter techniques, has also been reported (128).
POSTOPERATIVE CARE
Intraoperative and Immediate Postoperative Management
Cardiopulmonary Bypass (CPB)
Selected pediatric cardiac surgical procedures may be performed without the use of CPB such as modified Blalock-Taussig shunt or repair of coarctation of the aorta. However, the majority of cardiac operations performed require CPB. Thus, physicians caring for these patients must be familiar with the equipment and techniques used to perform CPB, its sequelae on end-organ function, and several intraoperative strategies used to minimize the associated morbidities. The primary function of CPB is to temporarily replace the major functions of the heart and lungs while surgical interventions are performed on these organs. A typical CPB circuit used to perform these functions includes venous cannula(s) that drain systemic venous blood from the vena cavae or systemic venous atrium, a reservoir, a heat exchanger, a membrane oxygenator, a roller pump, a filter, and an arterial cannula to return blood to the aorta. Before initiation of CPB, the circulated blood must be modified to provide an appropriate composition of oxygen, carbon dioxide, pH, temperature, hematocrit, oncotic pressure, electrolytes, and glucose (129). The circuit is “primed” with standardized quantities of crystalloid solution, albumin, mannitol, sodium bicarbonate, heparin, calcium, and packed red blood cells. The patient is anticoagulated with heparin for the duration of CPB time, and cooled to a variable extent to minimize metabolic needs and oxygen consumption. Because hypothermia causes increased viscosity and red cell rigidity, hemodilution is used during hypothermic cardiopulmonary bypass.
To obtain a motionless heart for intracardiac repairs, the aorta is cross-clamped and a potassium-rich cardioplegia solution is injected into the proximal ascending aorta. Asystole develops once the cardioplegia solution perfuses the coronary circulation. The combination of cardioplegia and hypothermia provides myocardial protection for several hours. Following placement of the aortic cross clamp, blood from aortopulmonary collaterals will continue to return to the left atrium. To eliminate the left atrial blood return and facilitate certain complex left heart operations, deep hypothermic circulatory arrest (DHCA) may be used. Deep hypothermia refers to cooling of the core temperature to 18°C to 20°C. During circulatory arrest the CPB pump is shut off. In addition to the absence of blood returning to the left atrium, the perfusion cannula may be removed from the surgical field, creating optimal conditions for an accurate repair. Due to concerns about neurologic outcome following DHCA, regional perfusion techniques have recently been designed to minimize or avoid the use of circulatory arrest (130). The neurologic sequelae of CPB and DHCA are discussed in the section on neurologic complications later in this chapter. Following rewarming, weaning and separation from CPB, protamine is administered to reverse the effect of heparin (129). Additional blood components may be required to control bleeding immediately following CPB.
A number of adverse effects from CPB will impact on the postoperative course. During CPB, formed elements of the blood are exposed to artificial surfaces and sheer stress. Ischemia reperfusion injury occurs, as does microembolization of gas bubbles and particulate matter. Hemodilution may cause problems with oxygen delivery and dilution of clotting factors. These events trigger a complex neurohumoral and inflammatory response. CPB is associated with the release of endogenous catecholamines, vasopressin, endothelin, and activation of the renin-angiotensin-aldosterone axis, all of which contribute to elevation of systemic and pulmonary vascular resistances and fluid retention (131,132,133,134,135,136,137,138,139,140,141,142). A generalized inflammatory response occurs following CPB, and the complement, coagulation and fibrinolytic systems are activated (143,144,145,146). Capillary leak also occurs, related to fluid retention, the inflammatory response, and dilution of plasma proteins. White blood cells and platelets are also activated, leading to additional release of inflammatory mediators and proteolytic enzymes (147,148,149). Pulmonary leukosequestration occurs, as does oxygen free radical generation (150,151). Platelet counts
fall following CPB, and clotting factors are diluted (140,152). Myocardial systolic dysfunction also occurs following CPB in infants and children (153). The end result of these processes is a clinical picture of end-organ dysfunction of variable severity. The cardiovascular, pulmonary, hematologic and renal systems tend to be the most severely affected. Myocardial dysfunction, elevated systemic and pulmonary vascular resistance, abnormal gas exchange and decreased pulmonary compliance, bleeding and fluid retention all may be present in the early postoperative period.
fall following CPB, and clotting factors are diluted (140,152). Myocardial systolic dysfunction also occurs following CPB in infants and children (153). The end result of these processes is a clinical picture of end-organ dysfunction of variable severity. The cardiovascular, pulmonary, hematologic and renal systems tend to be the most severely affected. Myocardial dysfunction, elevated systemic and pulmonary vascular resistance, abnormal gas exchange and decreased pulmonary compliance, bleeding and fluid retention all may be present in the early postoperative period.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


