Prenatal Diagnosis in the Molecular Age— Indications, Procedures, and Laboratory Techniques
Arie Drugan
Nelson B. Isada
Mark I. Evans
The modern era of molecular and biochemical genetics commenced with the observations of Sir Archibald Garrod at the beginning of the twentieth century. He proposed that four diseases—namely, alkaptonuria, albinism, cystinuria, and pentosuria—resulted from inherited disorders of chemical metabolism. He also suggested that these disorders, which he called “inborn errors of metabolism,” represented only a small fraction of every human’s “chemical individuality” that had gone awry (1).
Advances in biochemistry have confirmed Garrod’s concepts by characterizing the structural protein abnormality or enzymatic defect of many disorders. Other advances in molecular genetics have allowed precise identification of the defect in the deoxyribonucleic acid (DNA) message, sometimes before the protein defect itself is known (2). This knowledge has direct and immediate applications in the field of prenatal diagnosis (3). This chapter discusses gene organization; mutations and polymorphism analysis; molecular diagnostic techniques; DNA cloning; an approach to disorders diagnosable by molecular genetics; biochemical disorders not amenable to DNA technology or better studied by protein chemistry techniques; and carrier screening.
GENE ORGANIZATION
The Watson-Crick double-helix model of DNA organization is well known (4). DNA conveys information encoded by a series of four nucleotides—adenine (A), thymine (T), cytosine (C), and guanine (G)—that are connected sequentially on two strands. The two strands complement each other, with nucleotide base pairs (bp) being formed by hydrogen bonding between adenine-thymine and guanine-cytosine. Eukaryotic DNA is located in the nucleus and organized into structures called chromosomes. During interphase, chromosomes are not visible by light microscopy. They can be observed only when the genetic content has doubled and the chromosomes condense before mitosis. Chromosomal material is organized into euchromatin and heterochromatin. Euchromatin is vigorously transcribed into ribonucleic acid (RNA). Heterochromatin is relatively inactive. An example of heterochromatin is the inactivated X chromosome.
An unexpected discovery made in the 1970s was that some regions of the eukaryotic chromosome do not code for any known protein (5). Specifically, these noncoding regions (introns), were noted to be interspersed within coding regions (exons) (6,7). Exons carry information to direct the assembly of amino acids into a protein, whereas introns do not. Messenger RNA (mRNA) acts as an intermediate molecule to convey information encoded in the DNA by a process called translation. Posttranslational modification of mRNA takes place such that introns are removed and exons are joined together before amino acid sequences are formed. After additional biochemical modifications, the mRNA passes out of the nucleus into the cytoplasm, where protein-synthesizing organelles are located.
Approximately 60% of the human genome is comprised of regions of unique nucleotide sequences that presumably
code for proteins (8). It is estimated that there are 50,000 to 100,000 expressed genes and proteins active in humans; however, expressed genes comprise less than 10% of total genomic DNA. A significant portion of the human genome, perhaps approximately 40%, contains repetitive DNA sequences (9). Various terms are used for the different classes of repetitive DNA sequences found in humans. Highly repetitive sequences are found in the chromosome region adjacent to the centromere. These are simple sequences that are repeated thousands of times, are present in more than 104 copies, and comprise approximately 20% of the genome.
code for proteins (8). It is estimated that there are 50,000 to 100,000 expressed genes and proteins active in humans; however, expressed genes comprise less than 10% of total genomic DNA. A significant portion of the human genome, perhaps approximately 40%, contains repetitive DNA sequences (9). Various terms are used for the different classes of repetitive DNA sequences found in humans. Highly repetitive sequences are found in the chromosome region adjacent to the centromere. These are simple sequences that are repeated thousands of times, are present in more than 104 copies, and comprise approximately 20% of the genome.
Other repetitive, simple sequences are several hundred base pairs long, and are separated easily by centrifuging slightly fragmented DNA through a cesium chloride density gradient. DNA separated by this process is called satellite DNA, because the centrifuged DNA forms a main band and several satellite bands above and below the main band. In humans, four satellite bands comprise approximately 6% of the total DNA, each band representing tandem repeat sequences. Blocks of satellite DNA are readily localized by in situ hybridization to regions around the centromeres of metaphase chromosomes. Satellite DNA should not be confused with satellites, a cytogenetic term referring to the segment of an acrocentric chromosome distal to short arm and separated by a constriction.
Other classes of repetitive DNA include moderately repetitive DNA, which is gene size in length, repeated from 10 to 1,000 times, and comprises approximately 20% of genome (i.e., one-half of the 40% that is repetitive DNA); tandem repeat sequences, which are stretches of DNA in which a short nucleotide sequence is repeated 20 to 100 times, the exact number varying from person to person; alphoid DNA, which is a chromosome-specific, repeated, monomeric 170-bp unit located in centromeric regions; and Alu sequences, which are highly repetitive 300-bp sequences that are not clustered around centromeres, but are more evenly distributed throughout the genome and interspersed within longer stretches of unique or moderately repetitive DNA. Most contain a single cleavage site near the middle for the restriction enzyme Alu I, derived from the bacterium Arthrobacter luteus (see Restriction Fragment Length Polymorphism Analysis below). Almost 1 million Alu sequences are present in the human genome, accounting for 3% to 6% of the total DNA. Each individual human has a unique amount of repetitive DNA. This genetic fingerprint is used for paternity testing and forensic analysis.
A discovery involving repeat sequences demonstrates an association between changes in length of triplet repeats and human disease. A significant increase in length of the normal sequence of three repeated nucleotides is associated with marked tendency to clinical morbidity. This was first described in the fragile X syndrome, a common form of heritable male mental retardation, where a polymorphic sequence of -CGG- repeats on the X chromosome is increased from a mean of 29 repeats to more than 200 repeats in affected males. Triplet expansion of -GCT- is associated with congenital myotonic dystrophy. Triplet expansion of -CAG- is found in spinal-bulbar muscular atrophy, spinocerebellar ataxia, and Huntington disease (10,11,12).
Regulation of gene expression occurs at many levels. Substrate concentrations both enhance and repress specific enzymes, especially in bacteria. In eukaryotes and prokaryotes, a region separate from a given gene but still involved in regulation of expression is called promoter; it binds RNA polymerase and initiates transcription. Similar functional areas have been found that bind a large group of transcription factors. In eukaryotes, such an area or box contains a common or consensus nucleotide sequence, CAAT, and is called a CAAT box. A conserved area associated with RNA polymerase positioning contains repeated sequences of the nucleotides thymine and adenine and is called a TATA box.
Extranuclear DNA found in mitochondria (mtDNA) is inherited independently of nuclear DNA, is 16.5 kb in length, contains no introns, and is organized as a double-stranded circle. mtDNA is subject to a relatively high rate of mutation compared to nuclear DNA and is inherited maternally, the paternal spermatic mitochondria being excluded from the oocyte at the time of fertilization. A variety of neonatal and pediatric disorders, predominantly neuromuscular, are associated with mtDNA deletions (13,14,15).
ELEMENTS OF MOLECULAR GENETICS
The deciphering of DNA structure and the genetic code has advanced understanding of the molecular basis of genetic disease. Alterations in DNA sequence may affect a gene structure or function, and may result in an altered gene product that may (or may not) be expressed as genetic disease. An alteration in the sequence of nucleotides within a gene is termed a mutation.
Many types of mutations can affect a gene. The DNA within a gene contains information for the final sequence of a protein and signals for the correct expression and processing of mRNA. If the actual coding region is altered, then the resultant protein may be changed. These alterations can be in the form of deletions that may be many kilobases in length or as small as a single base, inversions or translocations that produce no net nucleotide changes but potential or actual protein changes, or single-base substitution. Even a change at the junction of a coding and noncoding region can result in abnormal mRNA formation. Defects in the promoter region may result in too little or too much expression of mRNA, which will be reflected in abnormal protein synthesis. A deletion of all or part of the gene will almost always result in the disruption of normal gene expression.
Two examples of molecular pathology can be seen in Ashkenazi Jews (16). In the severe infantile type of Tay-Sachs disease, a mutation at an exon-intron splice site in the α-subunit has been identified (i.e., G to C transversion); another mutation is a four-base insertion in exon 11 of the
α-subunit causing a frameshift mutation and marked reduction in mRNA (17,18). Another example is the F508 mutation, the most common (approximately 70%) mutation causing cystic fibrosis (CF) in people of northern European ancestry (19,20,21). The remaining 30% of CF cases in people of northern European ancestry are caused by a heterogeneous assortment of other mutations (i.e., W1282X). Commercial testing is available and can detect more than 90% of mutations in the northern European population (22). However, other ethnic groups carry their own particular repertoire of mutations and detection rate may be as low as 50% (23).
α-subunit causing a frameshift mutation and marked reduction in mRNA (17,18). Another example is the F508 mutation, the most common (approximately 70%) mutation causing cystic fibrosis (CF) in people of northern European ancestry (19,20,21). The remaining 30% of CF cases in people of northern European ancestry are caused by a heterogeneous assortment of other mutations (i.e., W1282X). Commercial testing is available and can detect more than 90% of mutations in the northern European population (22). However, other ethnic groups carry their own particular repertoire of mutations and detection rate may be as low as 50% (23).
A variety of approaches have been used to detect mutations. Optimally, determination of DNA structure and sequence, followed by elucidation of gene structure and organization in the normal allele, are completed before beginning a search for specific defects. However, only 5% to 10% of clinically significant mutations are a result of gross alterations in gene structure that are detectable by Southern blot analysis of genomic DNA, leaving unknown the remaining 90% to 95%. The problem is compounded by normal variation in the nucleotide sequence (polymorphism). Thus, when variations from the normal sequence are found, additional analysis is required before these changes can be construed as being a disease-causing mutation.
Because DNA is present in each cell nucleus, any nucleated cell theoretically is suitable for DNA analysis, regardless of whether the gene in question is being transcribed and expressed. Thus, leukocytes, amniocytes, and chorionic villi all are candidate cells for DNA analysis, using Restriction Fragment Length Polymorphism (RFLP). Other methods also used for DNA diagnosis include Southern blot, oligonucleotide probes, and polymerase chain reaction (PCR). Northern blotting is used for RNA analysis.
RFLP analysis uses bacterial enzymes that recognize and cleave DNA at specific sites. Presumably, these enzymes evolved as a defense mechanism against hostile, invading DNA, as might occur with bacteriophages. Because these cleavage sites are quite specific, that is, are restricted to specific palindromic sequences 4 to 10 nucleotides in length, these enzymes are called restriction endonucleases. When these enzymes are added to eukaryotic DNA, the resultant mixture contains a variety of DNA fragments of different sizes, which can be separated by gel electrophoresis and transferred for analysis by Southern blotting. Each enzyme cuts an individual’s DNA according to the positions of the cleavage sites, with every person having his or her own unique pattern of cleaved DNA fragments. Thus, people are polymorphic for the resulting lengths of DNA fragments. Many of these recognition-site polymorphisms are neutral and represent normal inherited variability. These characteristics have given rise to the term restriction fragment length polymorphisms, which refers to the polymorphic patterns observed in specific nucleotide sequences that are cleaved by bacterial restriction enzymes (Fig. 10-1). The resultant mixture of DNA fragments can be separated and further characterized by gel electrophoresis, Southern blotting, and oligonucleotide probes (24). DNA alterations that affect an RFLP site either by creating a new site for endonuclease cleavage or eliminating a previously existing one can be detected on Southern blot as a result of changes in the size of the DNA fragment associated with this site (25). If, by chance, either a mutation or normal sequence corresponds to an RFLP site, this situation can be exploited for allele identification by using linkage analysis.
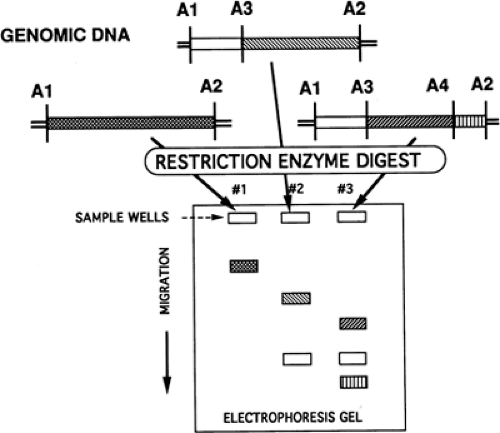 Figure 10-1 Restriction fragment length polymorphisms. Lane 1, A1, and A2 present; lane 2, A1-A3 present; lane 3, A1-A4 present; A1-A5, hypothetical polymorphisms. |
An initial use of such mapping involved the Huntington disease locus (26). Another early application was for prenatal identification of the sickle-cell mutation in the β chain of hemoglobin (27,28). For example, the restriction enzyme Dde I, derived from the bacterium Desulfovibrio desulfuricans aestuarii, recognizes the nucleotide sequence -CTNAG- (where N indicates that any nucleotide may occupy that position) that occurs within the hemoglobin A (-CTGAG-) and hemoglobin C (-CTAAG-) gene, but not within hemoglobin S (-CTGTG-). In hemoglobin S, the nucleotide thymine is substituted for adenine, which is not recognized by Dde I and thus is not cleaved by Dde I. This results in a much larger RFLP fragment that can be recognized on Southern blot. Initially, this technology used unamplified DNA and Southern blot transfer. Target gene sequences can now be preamplified a million-fold by PCR (see next page) and then cut by restriction endonucleases, which greatly facilitates target sequence recognition. This approach is potentially useful in prenatal diagnosis, particularly when the quantity of clinical material is limited.
When the precise nucleotide mutations of the abnormal gene causing the disease are unknown, linkage analysis is a very powerful technique that can be used for diagnosis by association of the diseased gene to a known gene or polymorphic site (29). This implies that the closer together two genetic traits are on a chromosome, the more likely they are to segregate together during meiosis and the less likely crossing-over occurs between them. When genes are in such close proximity that crossing-over rarely occurs, such as for
hemophilia A and color blindness, the two sites are said to be linked (30). Any polymorphism that is linked with the trait of interest is termed informative. Unfortunately, family studies are not always informative, because linked molecular or clinical polymorphisms are not always present.
hemophilia A and color blindness, the two sites are said to be linked (30). Any polymorphism that is linked with the trait of interest is termed informative. Unfortunately, family studies are not always informative, because linked molecular or clinical polymorphisms are not always present.
The likelihood that two given traits are linked can be described mathematically by the logarithm of the odds (LOD) score, developed in 1955 by Morton (31). It can be derived from recombination observed between clinical or biochemical traits from pedigree analysis, or from molecular polymorphisms (32). The probability of recombination during meiosis between two loci is quantified by the recombination fraction (i.e., theta or q), the maximum being 0.5. The LOD score is derived from various values of q. Viewed simplistically, the higher the LOD score, the higher the likelihood of linkage. Because this number is used on a logarithmic scale, each integer increase reflects a tenfold increase in likelihood of linkage. Thus, a LOD score of 4 suggests that there is linkage between two polymorphisms, the odds of random association being 10,000:1. A LOD score of zero suggests there is no linkage and that the two traits are on different chromosomes or are far apart on the same chromosome. Tight linkage indicates little or no recombination and suggests an actual physical proximity of two polymorphisms, measured in physical map distances, with the common unit of genetic distance reported as centimorgans (cM). Linkage disequilibrium describes closely linked genes that occur more frequently than would be expected from random distribution, suggesting nonrandom mating or some survival advantage from natural selection. Examples of linkage disequilibrium include the carrier states for sickle cell disease and thalassemia, where those affected have increased resistance to certain types of malarial infections.
Other methods used for DNA analysis include Southern blot, oligonucleotide probes, and PCR. Northern blot is used for RNA analysis. Southern blot is a standard method for DNA analysis in both the clinical and basic science settings. In the Southern blot technique, named after Edwin Southern, double-stranded DNA is digested by a restriction endonuclease chosen because of its ability to detect a DNA polymorphism (33). After endonuclease digestion, the resulting DNA fragments are separated using gel electrophoresis. The DNA in the gel is denatured to generate single-stranded DNA molecules. DNA fragments are transferred from the gel to nylon filter paper (blotting), and specific filter-bound DNA fragments then can be detected by hybridization. A radiolabeled DNA or RNA probe is used that has sequence homology to the DNA fragment of interest, usually 200 to 2,000 bases long. Subsequent autoradiography produces a radiographic film with banding patterns that indicate the hybridization locations on the filter that reflect the fragment sizes of the DNA sequences homologous to that particular probe (Fig. 10-2).
Allele-specific oligonucleotide hybridization has proven to be a valuable technique that measures the specific binding of short (18 to 20), labeled oligonucleotide probes, that match exactly either the wild-type) normal (or the mutant DNA sequence, under stringent washing conditions. Only the probes that exactly complement the immobilized DNA will remain bound and thus generate a signal seen on autoradiography. Conner (34) originally described this technique for the detection of sickle cell β-globin allele. This technique greatly facilitates the evaluation of genetic disorders in which the gene has to be screened for numerous mutations such as thalassemia, Tay-Sachs, Gaucher, or CF.
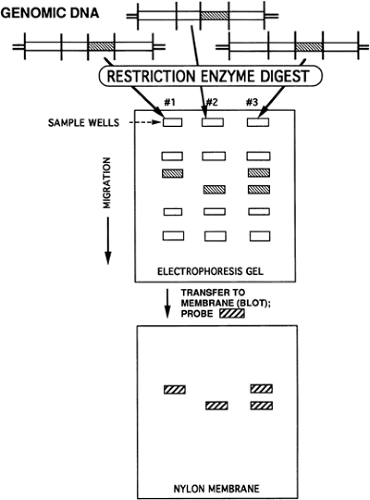 Figure 10-2 Southern blot for a hypothetical autosomal recessive disorder. Lane 1, unaffected; lane 2, affected; lane 3, carrier. |
PCR has revolutionized the field of molecular genetics (35,36). It’s discoverer, Kary Mullis, won the Nobel Prize in Chemistry in 1993. This procedure allows in vitro amplification of minute amounts of DNA to generate sufficient quantities of signal to make detection by more traditional methods possible. PCR makes use of Taq I, a relatively heat-stable bacterial enzyme derived from Thermus aquaticus, a thermoacidophilic bacterium. If the target nucleotide sequence is known, a specific set of oligonucleotides, called primers, can be synthesized to encompass the target sequence. The target DNA, oligonucleotide primers, Taq I polymerase, and free nucleotides are placed in solution. This reaction mixture is further heated to allow already denatured DNA to anneal with the oligonucleotides, between which the polymerase synthesizes complementary strands (Fig. 10-3). Repeated cycles of heating and cooling result in cyclic primer sequence synthesis, leading to annealing and amplification of the target sequence, because
each set of DNA strands gives rise to two additional sets of sequence templates in each cycle of the reaction. This process can be automated to allow 20 to 30 cycles, which can produce more than a million-fold duplication of the target sequence within hours. Modifications of this process can be performed to allow:
each set of DNA strands gives rise to two additional sets of sequence templates in each cycle of the reaction. This process can be automated to allow 20 to 30 cycles, which can produce more than a million-fold duplication of the target sequence within hours. Modifications of this process can be performed to allow:
Analysis of RNA.
Analysis of multiple DNA areas (multiplex PCR). This technique allows detection of more than 97% of deletions in Duchenne muscular dystrophy and all those of patients at risk for Becker muscular dystrophy (37).
Selective amplification of one strand instead of both (i.e., asymmetric PCR).
Simultaneous use of one primer set within another to increase specificity (i.e., nested PCR).
Simultaneous use of two different primer sets, one of which selects for a normal sequence and the other for a mutant sequence (i.e., competitive oligonucleotide priming).
Simultaneous use of a known amount of a second, easily identified target DNA to measure the amount of original DNA (i.e., semiquantitative PCR).
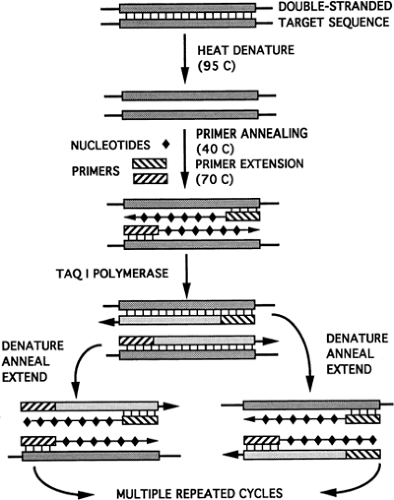 Figure 10-3 Polymerase chain reaction. |
Many technical difficulties must be addressed to eliminate both false-positive and false-negative PCR results. Problems with reagent or reactant contamination can lead to false-positive results and require that appropriate control methods be performed simultaneously to verify positive PCR results. Other problems, such as primer instead of target amplification and nonspecific amplification, must be recognized and avoided.
Northern blotting is used for RNA analysis. This requires prompt specimen processing and committed laboratory reagents and instruments because of ubiquitous ribonucleases, present even on finger surfaces. The general principles of the technique are similar to those for Southern blotting. Examination of the size and amount of a mRNA transcript is a useful initial step in evaluating the expression of mutated genes in cells or tissues. Fibroblasts and lymphocytes are good sources of mRNA. Hepatic or muscle tissue is also useful, if available. Placental tissue can be used if maternal cell contamination can be avoided. Of the cell’s total RNA, only 1% to 2% is mRNA, which is highly unstable at room temperature because of tissue RNA-ases. The remainder of the RNA is mainly ribosomal RNA (rRNA) and transfer RNA (tRNA). Once isolated, the mRNA is denatured, separated by agarose gel electrophoresis, transferred to a membrane filter, and analyzed by hybridization of a specific fluorescent or radiolabeled probe.
For most diseases studied at the molecular level, 5% to 10% of patients have no detectable mRNA for the gene product in question; 10% to 20% have reduced but detectable amounts of normal mRNA; and approximately 5% have some alteration in mRNA size. The approximately 50% remaining have normal amounts of normal-size mRNA. Given this information, it is possible to deduce the general type of mutation at the DNA level, such as large or total gene deletions, which are suggested by a total absence of mRNA. Mutations in promoter regions are suggested by reduced amounts of normal mRNA. Mutations at exon-intron junctions are suggested by changes in mRNA size—normal-size mRNA but abnormal function of protein hints at point mutations.
In some instances, the quantity of RNA is insufficient to be detected in the previously mentioned methods. In these cases, reverse transcriptase PCR methodology permits identification and isolation of small quantities of mRNA and thereby analyze genes in a more fastidious manner. Grompe et al. (38) were unable to detect by Northern analysis the mRNA in an ornithine transcarbamylase-deficient patient. Nonetheless, the mRNA was isolated and successfully amplified (after synthesis of a single-stranded complementary DNA [cDNA] by using the RNA-directed DNA polymerase, reverse transcriptase), and the mutation responsible for the disease was identified. This technique also allows isolation of the shorter cDNA fragments corresponding to the coding region of the gene of interest. As in Menkes’ (“kinky hair”) disease, a neurodegenerative disorder associated with a disturbance of copper metabolism, exon splicing is the characteristic result of the splice-junction mutations seen in this rather large gene (39). By using reverse transcriptase PCR, one can discern which exons are lacking by the size of fragment seen on agarose gel or by sequence analysis.
The basic steps in the study of a gene start with the isolation of a DNA molecule complementary to its mRNA, called cDNA, that contains only exonic sequences. The first
step in cDNA cloning is the isolation of mRNA from a particular cell or tissue that contains a significant amount of the desired mRNA. A retroviral enzyme, reverse transcriptase, is used to synthesize cDNA from the mRNA. Because the cell or tissue mRNA contains transcripts of many genes, the resultant pool of cDNA will be heterogeneous and must be sorted out. The cDNA strands are inserted into a vector to form a cDNA library. A second type of chromosome library is composed of fragments of native genomic DNA and contains introns and other noncoding regions. Vectors include viruses that can replicate within bacteria, such as bacteriophage lambda, or autonomous, self-replicating, circular DNA molecules found in bacteria, called plasmids. Another vector that combines properties of plasmids and bacteriophage lambda are called cosmids. Cosmids are plasmid vectors into which larger fragments of DNA can be cloned. The term cosmid is derived from the presence of internal cohesive end sites (cos) that have been inserted into a plasmid. Cos are nucleotide sequences from bacteriophage lambda between which DNA sequences are normally expressed as capsule proteins. In cosmids, these sequences can be replaced with other nucleotide sequences, which then can be expressed and concentrated in vitro.
step in cDNA cloning is the isolation of mRNA from a particular cell or tissue that contains a significant amount of the desired mRNA. A retroviral enzyme, reverse transcriptase, is used to synthesize cDNA from the mRNA. Because the cell or tissue mRNA contains transcripts of many genes, the resultant pool of cDNA will be heterogeneous and must be sorted out. The cDNA strands are inserted into a vector to form a cDNA library. A second type of chromosome library is composed of fragments of native genomic DNA and contains introns and other noncoding regions. Vectors include viruses that can replicate within bacteria, such as bacteriophage lambda, or autonomous, self-replicating, circular DNA molecules found in bacteria, called plasmids. Another vector that combines properties of plasmids and bacteriophage lambda are called cosmids. Cosmids are plasmid vectors into which larger fragments of DNA can be cloned. The term cosmid is derived from the presence of internal cohesive end sites (cos) that have been inserted into a plasmid. Cos are nucleotide sequences from bacteriophage lambda between which DNA sequences are normally expressed as capsule proteins. In cosmids, these sequences can be replaced with other nucleotide sequences, which then can be expressed and concentrated in vitro.
An alternative approach to cDNA cloning uses mRNA itself instead of cDNA. Using mRNA is advantageous because the mRNA pool, although heterogeneous, is not nearly so complex as a corresponding pool of genomic DNA fragments that contains a mixture of introns, nonexpressed genes, and DNA from noncoding regions. Methods of cDNA cloning to detect the presence of a specific cDNA segment include screening a cDNA library with synthetic oligonucleotide probes and screening a bacteriophage lambda library with antibodies directed against the protein of interest. Using the first approach, a set of radiolabeled oligonucleotide probes is synthesized with sequences complementary to those predicted for the mRNA from the amino acid sequence found in the protein. These oligonucleotides are used to probe a cDNA library that contains bacterial colonies infected with bacteriophage lambda. With the second approach, antibodies against the protein of interest are used to detect a corresponding protein expressed by cDNA inserted into bacteriophage lambda.
cDNA for many genes have been cloned using these approaches. Nucleotide sequencing is the first step in the analysis of a cDNA. With this information, the investigator can identify potential sites of restriction endonuclease cleavage, can compare the deduced amino acid sequence with that of the protein of interest, and can identify regions of homology with other proteins, which may suggest evolutionary relationships and previously unrecognized functions. In addition to providing nucleotide and amino acid sequence information, these cDNA can be used as hybridization probes to characterize and quantitate corresponding mRNA in cells and tissues from patients with a variety of diseases.
The process by which DNA analysis leads to an identifiable protein abnormality was previously called “reverse genetics”; it is now called “positional cloning” (40,41). Several hundred genes associated with human disease have been identified. In some diseases, such as Cystic Fibrosis, the triple-nucleotide/single-amino-acid deletion in the CFTR protein has been recognized. For other diseases, the gene and protein are better characterized, such as for neurofibromatosis type 1 (42). For most human diseases, however, the genes and the defect in protein synthesis have yet to be defined.
PRENATAL DIAGNOSIS
Whether by cytogenetic, biochemical, or molecular methods, prenatal diagnosis of an affected fetus requires obtaining either fetal tissue (i.e., blood) or other tissue for analysis that is representative of the fetus (e.g., amniocytes or placenta). Invasive procedures for prenatal diagnosis of fetal disease are available throughout gestation, from the first trimester onward. Even earlier, assisted reproduction technologies (ART) enable diagnosis (or exclusion) of several disorders on the 4- to 8-cell embryo before implantation.
Invasive procedures for diagnosis in pregnancy of fetal genetic disorders have been available for almost four decades, since the introduction of techniques for culturing and karyotyping of amniotic fluid fibroblasts in the mid-1960s (43). The first diagnosis of a fetal chromosome anomaly by amniocentesis (44) was followed shortly by the diagnosis of an enzyme deficiency in amniotic fluid cells (45). Thereafter, collaborative studies established the safety and accuracy of midtrimester amniocentesis, so that this technique became a routine part of prenatal care in high-risk patients and the gold standard against which other procedures for prenatal diagnosis are compared (46,47).
Despite its proven efficacy, a major disadvantage of amniocentesis is the availability of results late in the second trimester, generally 18 to 20 weeks of gestation. The emotional and physical implications of termination of pregnancy this late in gestation are obvious. Improvement in ultrasonography machinery and increasing expertise in ultrasound-guided procedures enabled physicians in the late 1980s to attempt prenatal diagnosis in the first trimester, introducing chorionic villus sampling (CVS) and early amniocentesis. These technical developments are backed and reinforced by increasing preference on the part of patients for first trimester prenatal diagnosis (48). Chorionic villus sampling is usually performed between 11 and 13 weeks of gestation so that results are available by the end of the first trimester. The accuracy and safety of CVS are quite comparable to those of mid trimester amniocentesis (49,50) and the early results allow patients privacy in reproductive decisions and an earlier and safer termination of pregnancy if so opted. An alternative to CVS was offered by early amniocentesis performed between 10 and 14 weeks of gestation, but this technique has been largely abandoned (51).
Over the years, a change in the pattern of indications for prenatal diagnosis has been observed. The most common
indication for genetic counseling and prenatal diagnosis is the need to evaluate the karyotype of the fetus, this being indicated in more than 70% of cases of advanced maternal age (defined as 35 years or older at birth). Other “classical” indications to evaluate fetal karyotype include a previous, affected offspring and a balanced structural rearrangement of parental chromosomes, the latter being clinically evident as recurrent pregnancy loss (Table 10-1). In recent years, increased use of biochemical serum screening and of ultrasonographic screening for fetal chromosome anomalies have caused more young patients, previously considered to be at low risk for fetal aneuploidy, to opt for invasive prenatal testing. The combination of “double,” “triple,” or “quadruple” serum screening (α-fetoprotein [AFP], human chorionic gonadotropin [hCG], and unconjugated estriol [uE3], with or without inhibin A) and maternal age will select for prenatal testing a sub-group of patients among whom 65% to 75% of chromosomally abnormal conceptions will be contained. Using a risk cutoff for fetal aneuploidy equal to that of age 35 years, some 5% of young pregnant patients will have a positive screening test, and 1 in 50 amniocenteses performed for this indication will diagnose a chromosomally abnormal conception (52). In the second trimester, sonographic markers for fetal chromosome anomalies are observed in 3% to 5% of pregnancies (53) (Table 10-2) and are another indication for fetal karyotyping. The most worrisome of these findings are abnormalities of fetal neck, indicating the need for evaluation of fetal chromosomes even in association with normal biochemical serum screening in young patients (54).
indication for genetic counseling and prenatal diagnosis is the need to evaluate the karyotype of the fetus, this being indicated in more than 70% of cases of advanced maternal age (defined as 35 years or older at birth). Other “classical” indications to evaluate fetal karyotype include a previous, affected offspring and a balanced structural rearrangement of parental chromosomes, the latter being clinically evident as recurrent pregnancy loss (Table 10-1). In recent years, increased use of biochemical serum screening and of ultrasonographic screening for fetal chromosome anomalies have caused more young patients, previously considered to be at low risk for fetal aneuploidy, to opt for invasive prenatal testing. The combination of “double,” “triple,” or “quadruple” serum screening (α-fetoprotein [AFP], human chorionic gonadotropin [hCG], and unconjugated estriol [uE3], with or without inhibin A) and maternal age will select for prenatal testing a sub-group of patients among whom 65% to 75% of chromosomally abnormal conceptions will be contained. Using a risk cutoff for fetal aneuploidy equal to that of age 35 years, some 5% of young pregnant patients will have a positive screening test, and 1 in 50 amniocenteses performed for this indication will diagnose a chromosomally abnormal conception (52). In the second trimester, sonographic markers for fetal chromosome anomalies are observed in 3% to 5% of pregnancies (53) (Table 10-2) and are another indication for fetal karyotyping. The most worrisome of these findings are abnormalities of fetal neck, indicating the need for evaluation of fetal chromosomes even in association with normal biochemical serum screening in young patients (54).
TABLE 10-1 INDICATIONS FOR PRENATAL DIAGNOSIS | |
|---|---|
|
TABLE 10-2 RELATIVE RISK FOR ANEUPLOIDY ASSOCIATED WITH ISOLATED SONOGRAPHIC MARKERS | |||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||
The most effective screening test for Down syndrome is probably the integrated test, based on estimation of the nuchal translucency on ultrasonography, hCG, and pregnancy-associated plasma protein A (PAPP-A) in the first trimester, combined with AFP, HCG, estriol, and inhibin A in the second trimester of pregnancy. The integrated risk assessment is reported to have a 94% detection rate and a 5% false-positive rate (55). The results of biochemical or ultrasonography screening also can be used to modify the risk of aneuploidy in the population previously considered at risk, reducing the number of invasive diagnostic procedures in patients of advanced maternal age by more than half (56,57). Furthermore, “at-risk” patients who previously declined amniocentesis may be influenced to accept invasive prenatal diagnosis following a positive screen result (57). However, a major problem of this “integrated” approach is that results obtained in the first trimester are withheld from the patient, and the advantages of first trimester diagnosis are lost.
The need for rapid karyotyping may arise when fetal anomalies are suspected near the statutory limit for termination
of affected pregnancies, that is, after an abnormal result on biochemical or ultrasonography evaluation. In those cases, the diagnostic options are “late” CVS (58) or cordocentesis and karyotyping of fetal blood lymphocytes. Other tests that may be performed on blood obtained by cordocentesis include hematologic parameters, acid-base balance, and immunologic status of the fetus (59).
of affected pregnancies, that is, after an abnormal result on biochemical or ultrasonography evaluation. In those cases, the diagnostic options are “late” CVS (58) or cordocentesis and karyotyping of fetal blood lymphocytes. Other tests that may be performed on blood obtained by cordocentesis include hematologic parameters, acid-base balance, and immunologic status of the fetus (59).
Birth of a child with an inherited genetic disorder caused by malfunction in a single gene implies a 25% to 50% risk of recurrence in subsequent gestations, depending on the specific mode of inheritance of that disease. This risk and the associated genetic burden of the disease may be considered so high that many couples will opt to avoid further reproduction unless prenatal diagnosis is available. Fetal karyotype is uninformative in these cases. Prenatal diagnosis of mendelian disease is performed using biochemical or molecular techniques on fetal or placental tissue. Biochemical assays include assessment of gene products such as enzymes, receptors, and transport proteins, and metabolites such as amino acids, organic acids, vitamins, and hormones. When the underlying biochemical defect is known and is expressed in accessible fetal tissue or cells, prenatal diagnosis can be achieved by enzyme analysis of material obtained by CVS, amniocentesis, or cordocentesis. Because variability caused by different mutations and different genomic backgrounds exists among families, additional testing of leukocytes or cultured skin fibroblasts from presumably unaffected parents and siblings can provide valuable information. In addition to the benefit in interpretation of prenatal results, such studies may provide a reliable means for identification of other carriers among members of the extended family.
Prenatal diagnosis is now available for many inherited metabolic disorders. For an autosomal recessive disease, biochemical assays can be used should discriminate among homozygous affected, heterozygous unaffected, and homozygous normal fetuses. Assays for detection of autosomal dominant diseases, such as some of the porphyrias, usually are capable of identifying affected homozygotes, but sometimes fail to differentiate conclusively affected heterozygotes from unaffected fetuses. Heterozygote detection in X-linked disorders is difficult because of the random X inactivation occurring in every pregnancy with a female fetus. Depending on the ratio of an active mutant X to the normal X in tissues involved in the pathogenesis of the disease, a female heterozygous for an X-linked disorder may be clinically normal, or may have mild or even severe disease manifestations (60). To complicate matters further, measured enzymatic activities also vary depending on the ratio of mutant to normal X chromosomes that are active in the analyzed specimen—chorionic villi, for example. Occasionally, the activity levels in chorionic villi will not correlate with clinical expression. Males, conversely, have only one X chromosome and are either hemizygous affected with deficient enzyme activity or hemizygous normal with activity in the normal range. Thus, prenatal biochemical assessment of X-linked disorders is less complicated if the fetus is male.
The use of direct and cultured fetal specimens for prenatal evaluation of metabolic disorders ideally requires the availability of normal control preparations. Except for trophoblasts and amniotic fluid cells that can be maintained in culture, availability of fresh controls is often a problem, and in most instances long-term frozen controls with partial loss of activity must be used. There are other potential pitfalls that seem specific for each of these tissue, cell, and fluid types. All samples should be analyzed as soon as possible, except those requiring initial tissue culture. Chorionic villi, fetal tissue biopsies, cell pellets, amniotic fluid supernatant, and fetal serum or plasma that are not used for tissue culture can be kept frozen and shipped on dry ice. Cell and tissue cultures, however, should be shipped at room temperature. Whenever possible, appropriate controls matched by gestational age should accompany the samples to be analyzed. Extraction and analysis of labile enzymes is especially difficult, because test results are very sensitive with respect to the duration of homogenization or sonication. Using fresh chorionic villi or amniocytes or freshly harvested trophoblasts helps preserve the activity of such labile enzymes (61).
Fetal liver and muscle biopsy should be considered only in the absence of other alternatives, because the risk for pregnancy loss associated with these invasive procedures is significantly higher. Fetal muscle biopsy has been used in rare cases of Duchenne muscular dystrophy (DMD), when molecular analysis of trophoblasts, amniocytes, or fetal leukocytes is nondiagnostic and family studies are uninformative. An in utero fetal muscle biopsy can be performed in the middle of the second trimester to assess dystrophin levels in myoblasts by in situ hybridization (62). Absence of dystrophin suggests an affected fetus.
Fetal liver biopsy also can be performed for certain rare enzyme deficiencies. For example, in one type of glycogenosis, glucose-6-phosphatase is decreased; this enzyme is expressed only in fetal liver and kidney. In the absence of direct DNA techniques, the only option available for prenatal diagnosis is fetal liver biopsy in which glucose-6-phosphatase activity can be measured. Fetal liver biopsy also is applicable in rare cases of ornithine transcarbamylase deficiency where family studies are uninformative and known deletions cannot be detected (63). In the future, it is probable that most of these procedures will be considered obsolete, and prenatal diagnosis of most genetic disorders will be performed by molecular analysis, including some mitochondrial disorders that may be amenable to prenatal diagnosis (64).
Prenatal Diagnosis in the First Trimester
Chorionic Villus Sampling
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


