Chapter 478 Platelet and Blood Vessel Disorders
Megakaryopoiesis
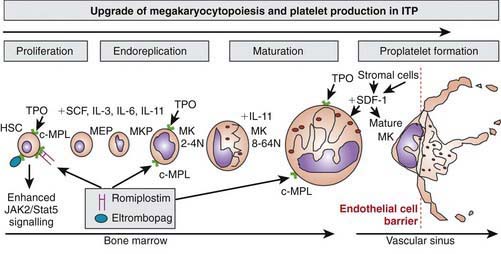
Platelets are non-nucleated cellular fragments produced by megakaryocytes within the bone marrow and other tissues. Megakaryocytes are large polyploid cells. When the megakaryocyte approaches maturity, budding of the cytoplasm occurs and large numbers of platelets are liberated. Platelets circulate with a life span of 10-14 days. Thrombopoietin (TPO) is the primary growth factor that controls platelet production (Fig. 478-1). Levels of TPO appear to correlate inversely with platelet number and megakaryocyte mass. Levels of TPO are highest in the thrombocytopenic states associated with decreased marrow megakaryopoiesis and may be variable in states of increased platelet production.
Thrombocytopenia
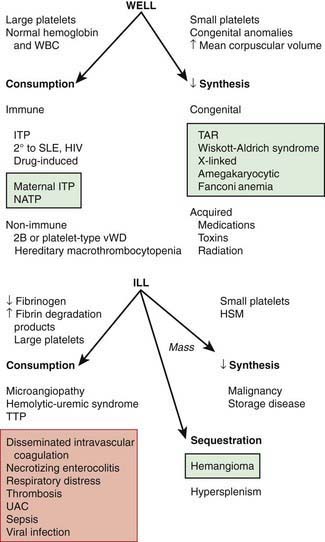
The normal platelet count is 150-450 × 109/L. Thrombocytopenia refers to a reduction in platelet count to <150 × 109/L. Causes of thrombocytopenia include: (1) decreased production on either a congenital or an acquired basis; (2) sequestration of the platelets within an enlarged spleen or other organ; and (3) increased destruction of normally synthesized platelets on either an immune or a nonimmune basis (Chapter 469; Tables 478-1 and 478-2 and Fig. 478-2).
Table 478-1 DIFFERENTIAL DIAGNOSIS OF THROMBOCYTOPENIA IN CHILDREN AND ADOLESCENTS
DESTRUCTIVE THROMBOCYTOPENIAS
Primary Platelet Consumption Syndromes
Combined Platelet and Fibrinogen Consumption Syndromes
IMPAIRED PLATELET PRODUCTION
SEQUESTRATION
HIV, human immunodeficiency virus; ITP, immune thrombocytopenic purpura; VWD, von Willebrand disease.
From Wilson DB: Acquired platelet defects. In Orkin SH, Nathan DG, Ginsburg D, et al, editors: Nathan and Oski’s hematology of infancy and childhood, ed 7, Philadelphia, 2009, WB Saunders, p 1555, Box 33-1.
Table 478-2 CLASSIFICATION OF FETAL AND NEONATAL THROMBOCYTOPENIAS*
| CONDITION | |
| Fetal | Alloimmune thrombocytopenia |
| Congenital infection (e.g., CMV, toxoplasma, rubella, HIV) | |
| Aneuploidy (e.g., trisomy 18, 13, or 21, or triploidy) | |
| Autoimmune condition (e.g., ITP, SLE) | |
| Severe Rh hemolytic disease | |
| Congenital/inherited (e.g., Wiskott-Aldrich syndrome) | |
| Early-onset neonatal (<72 hr) | Placental insufficiency (e.g., PET, IUGR, diabetes) |
| Perinatal asphyxia | |
| Perinatal infection (e.g., Escherichia coli, GBS, Haemophilus influenzae) | |
| DIC | |
| Alloimmune thrombocytopenia | |
| Autoimmune condition (e.g., ITP, SLE) | |
| Congenital infection (e.g., CMV, toxoplasma, rubella, HIV) | |
| Thrombosis (e.g., aortic, renal vein) | |
| Bone marrow replacement (e.g., congenital leukemia) | |
| Kasabach-Merritt syndrome | |
| Metabolic disease (e.g., proprionic and methylmalonic acidemia) | |
| Congenital/inherited (e.g., TAR, CAMT) | |
| Late-onset neonatal (>72 hr) | Late-onset sepsis |
| NEC | |
| Congenital infection (e.g., CMV, toxoplasma, rubella, HIV) | |
| Autoimmune | |
| Kasabach-Merritt syndrome | |
| Metabolic disease (e.g., proprionic and methylmalonic acidemia) | |
| Congenital/inherited (e.g., TAR, CAMT) |
CAMT, congenital amegakaryocytic thrombocytopenia; CMV, cytomegalovirus; DIC, disseminated intravascular coagulation; GBS, group B streptococcus; ITP, idiopathic thrombocytopenic purpura; IUGR, intrauterine growth restriction; NEC, necrotizing enterocolitis; PET, preeclampsia; SLE, systemic lupus erythematosus; TAR, thrombocytopenia with absent radii.
* The most common conditions are shown in bold.
From Roberts I, Murray NA: Neonatal thrombocytopenia: causes and management, Arch Dis Child Fetal Neonatal Ed 88:F359–F364, 2003.
Nachman RL, Rafii S. Platelets, petechiae, and preservation of the vascular wall. N Engl J Med. 2008;359:1261-1270.
Newman PK, Newman DK. Platelets and the vessel wall. In: Orkin SH, Nathan DG, Ginsberg D, et al, editors. Nathan and Oski’s hematology of infancy and childhood. ed 7. Philadelphia: Saunders Elsevier; 2009:1379-1399.
478.1 Idiopathic (Autoimmune) Thrombocytopenic Purpura
Pathogenesis
Why some children develop the acute presentation of an autoimmune disease is unknown. The exact antigenic target for most such antibodies in most cases of childhood acute ITP remains undetermined. although in chronic ITP most patients demonstrate antibodies against the platelet glycoprotein complexes, α11b-B3 and GPIb. After binding of the antibody to the platelet surface, circulating antibody-coated platelets are recognized by the Fc receptor on splenic macrophages, ingested, and destroyed. Most common viruses have been described in association with ITP, including Epstein-Barr virus (Chapter 246) and HIV (Chapter 268). Epstein-Barr virus-related ITP is usually of short duration and follows the course of infectious mononucleosis. HIV-associated ITP is usually chronic. In some patients ITP appears to arise in children infected with Helicobacter pylori or rarely following the measles, mumps, rubella vaccine.
Clinical Manifestations
Laboratory Findings
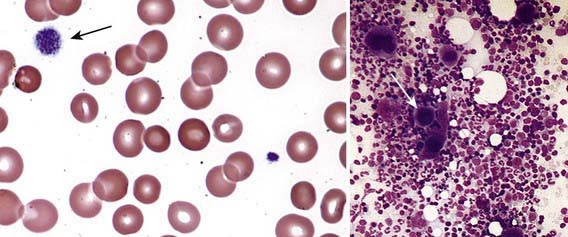
Severe thrombocytopenia (platelet count <20 × 109/L) is common, and platelet size is normal or increased, reflective of increased platelet turnover (Fig. 478-3). In acute ITP, the hemoglobin value, white blood cell (WBC) count, and differential count should be normal. Hemoglobin may be decreased if there have been profuse nosebleeds or menorrhagia. Bone marrow examination shows normal granulocytic and erythrocytic series, with characteristically normal or increased numbers of megakaryocytes. Some of the megakaryocytes may appear to be immature and are reflective of increased platelet turnover. Indications for bone marrow aspiration/biopsy include an abnormal WBC count or differential or unexplained anemia as well as findings on history and physical examination suggestive of a bone marrow failure syndrome or malignancy. Other laboratory tests should be performed as indicated by the history and physical examination. In adolescents with new-onset ITP, an antinuclear antibody test should be done to evaluate for SLE. HIV studies should be done in at-risk populations, especially sexually active teens. Platelet antibody testing is seldom useful in acute ITP. A direct antiglobulin test (Coombs) should be done if there is unexplained anemia to rule out Evans syndrome (autoimmune hemolytic anemia and thrombocytopenia) (Chapter 458) or before instituting therapy with IV anti-D.
Diagnosis/Differential Diagnosis
The well-appearing child with moderate to severe thrombocytopenia, an otherwise normal complete blood cell count (CBC), and normal findings on physical examination has a limited differential diagnosis that includes exposure to medication that induces drug-dependent antibodies, splenic sequestration due to previously unappreciated portal hypertension, and rarely, early aplastic processes, such as Fanconi anemia (Chapter 462). Other than congenital thrombocytopenia syndromes (Chapter 478.8), such as thrombocytopenia-absent radius (TAR) syndrome and MYH9-related thrombocytopenia, most marrow processes that interfere with platelet production eventually cause abnormal synthesis of red blood cells (RBCs) and WBCs and therefore manifest diverse abnormalities on the CBC. Disorders that cause increased platelet destruction on a nonimmune basis are usually serious systemic illnesses with obvious clinical findings (e.g., hemolytic-uremic syndrome [HUS], disseminated intravascular coagulation [DIC]) [see Table 477-1 and Fig. 478-2]. Isolated enlargement of the spleen suggests the potential for hypersplenism owing to either liver disease or portal vein thrombosis. Autoimmune thrombocytopenia may be an initial manifestation of SLE, HIV infection, common variable immunodeficiency, or rarely lymphoma. Wiskott-Aldrich syndrome (WAS; Chapter 120.2) must be considered in young males found to have thrombocytopenia with small platelets, particularly if there is a history of eczema and recurrent infection.



