Pharmacogenetics, Pharmacogenomics, and Pharmacoproteomics
J. Steven Leeder
Introduction
It is readily accepted that genetic factors play an important role in influencing a child’s potential physical characteristics such as height, weight, or hair color. Genetic factors are also important (although not sole) determinants of inter- and intraindividual variability in susceptibility to pediatric diseases as well as in the disposition of and response to medications used to treat those diseases. Pharmacotherapy in adults has benefited from knowledge of pharmacogenetic principles acquired over the past 50 to 60 years, but application to pediatric therapeutics is still in its infancy. On April 14, 2003, the International Human Genome Sequencing Consortium announced successful completion of the Human Genome Project initiated in 1990, 2 years after being presented in draft form (1). There is considerable hope that morbidity and mortality will be decreased through the development of more effective strategies to diagnose, treat, and prevent human disease, and society has every right to expect that children and adults will benefit equally from this investment of public funds. However, children present unique challenges in this context since developmental changes in drug disposition and response are superimposed upon a basal level of pharmacogenetic variability. The purpose of this chapter is to introduce the concepts of pharmacogenetics, pharmacogenomics, and pharmacoproteomics (and other “-omic” fields of endeavor spawned in the genome era) in the context of the changes in growth and development characteristic of the pediatric population.
Historical Considerations
In 1841, Alexander Ure reported that hippuric acid was formed from benzoic acid in the body leading physiological chemists to discover that many foreign substances excreted by humans were chemically altered relative to the forms that had been administered—the process we now refer to as drug biotransformation (or less properly, drug metabolism). At the beginning of the 20th century, Archibald Garrod proposed that enzymes were implicated in the detoxification of foreign substances. A key element of his later work was the concept that disproportionate responses to foreign substances could result from deficiency of the required detoxifying enzyme. A variation in the theme of altered responses to foreign substances (xenobiotics) became apparent with the synthesis of phenylthiocarbamide in 1931 when, in the process searching out artificial sweeteners, A. L. Fox discovered that some people found the chemical intensely bitter whereas others found it tasteless (2). It was not until the 1950s, however, that certain adverse drug reactions, such as unusually prolonged respiratory muscle paralysis due to succinylcholine, hemolysis associated with antimalarial therapy, and isoniazid-induced neurotoxicity, were recognized to be a consequence of inherited variation in enzyme activities as reviewed by Arno Motulsky in 1957 (3).
In 1959, Fridriech Vogel coined the term pharmacogenetics to describe the study of genetically determined variations in drug response and the first book on the subject was published in 1962 by Werner Kalow (4). Through a series of twin studies conducted during the late 1960s and early 1970s, Elliott Vesell illustrated the importance of genetic variation in drug disposition by observing that the half-lives of several drugs were more similar in monozygotic twins than in dizygotic twins (5). With the discovery of the debrisoquine/sparteine hydroxylase polymorphism (6,7), due to inherited defects in the cytochrome P450 2D6 gene (CYP2D6), and mephenytoin hydroxylase deficiency (CYP2C19) (8) in the late 1970s and early 1980s, the importance of genetic polymorphisms in drug-metabolizing enzymes has become increasingly apparent. This is particularly true in recent years due to an enhanced awareness of the number of clinically useful drugs that are
metabolized by polymorphically expressed enzymes and the proportion of treated patients who are affected.
metabolized by polymorphically expressed enzymes and the proportion of treated patients who are affected.
In the 1970s, an increasing appreciation of genetic influences on variability in drug disposition and response was accompanied by heightened awareness that environmental factors (e.g., diet, smoking status, and concomitant drug or toxicant exposure), physiologic variables (e.g., age, gender, disease, and pregnancy), and patient compliance also played important roles. Advances in analytic tools to accurately measure drugs and drug metabolites in biological fluids and the development of mathematical models to characterize and predict changes in drug concentration over time (pharmacokinetics) led to the application of pharmacokinetic principles to optimize drug therapy in individual patients. Introduction of therapeutic drug monitoring programs was the first application of personalized medicine—recognition that all patients were unique and that utilizing serum concentration–time data for an individual patient theoretically could be used to optimize pharmacotherapy was a significant advance over the concept of “one dose fits all.” However, routine therapeutic drug monitoring does not necessarily translate to improved patient outcome in all situations (9).
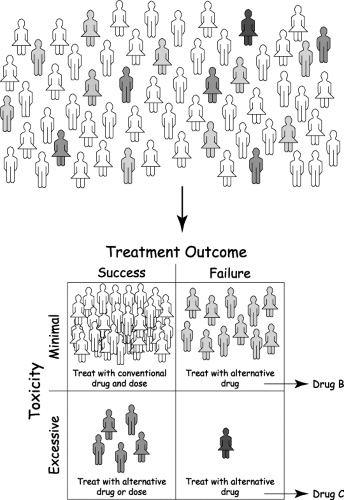 Figure 5.1. The promise of genomic medicine to human health and disease. The goal of personalized medicine will be achieved by identifying subgroups of patients who will respond favorably to a given drug with a minimum of side effects, as well as those who will not respond or who will show excessive toxicity with standard doses. A further benefit of pharmacogenomics will be the ability to select the most appropriate alternative drug for those patients who fail treatment with conventional drugs and doses. |
At a molecular level, the pharmacokinetic properties of a drug are determined by the genes that control its disposition in the body (e.g., absorption, distribution, metabolism, and excretion) with drug-metabolizing enzymes and drug transporters assuming particularly important roles. Over the past 25 years, the functional consequences of genetic variation in several drug-metabolizing enzymes have been described in subjects representative of different ethnic groups (10). Whereas the most common clinical
manifestation of pharmacogenetic variability in drug biotransformation is an increased risk of concentration-dependent toxicity due to reduced clearance and drug accumulation, it has become more apparent in recent years that the concentration–effect relationship (pharmacodynamics) is more relevant for optimizing drug efficacy. Therefore, the pharmacogenetics of drug receptors and other target proteins involved in signal transduction or disease pathogenesis can also be expected to contribute significantly to interindividual variability in drug response (11,12). However, the most important concept is that the pharmacogenetic determinants of drug response involve multiple genes and therefore, for a particular individual, polymorphisms in a single gene are unlikely to be predictive of response.
manifestation of pharmacogenetic variability in drug biotransformation is an increased risk of concentration-dependent toxicity due to reduced clearance and drug accumulation, it has become more apparent in recent years that the concentration–effect relationship (pharmacodynamics) is more relevant for optimizing drug efficacy. Therefore, the pharmacogenetics of drug receptors and other target proteins involved in signal transduction or disease pathogenesis can also be expected to contribute significantly to interindividual variability in drug response (11,12). However, the most important concept is that the pharmacogenetic determinants of drug response involve multiple genes and therefore, for a particular individual, polymorphisms in a single gene are unlikely to be predictive of response.
In 1987, the term “genomics” was introduced to describe the study of the structure and function of the entire complement of genetic material—the genome—including chromosomes, genes, and DNA (13). In 1990, the Human Genome Project was initiated as a $3 billion public investment with the goal of sequencing the entire complement of human genes by the year 2005 but more importantly, with the expectation that decreased morbidity and mortality through the development of more effective strategies to diagnose, treat, and prevent human disease would be the return on that investment (Fig. 5.1). The publicly funded initiative was forced to accelerate its efforts when J. Craig Venter announced that his company, Celera Genomics, would sequence the human genome first. The two efforts resulted in simultaneous publication of initial draft sequences in 2001 (1,14), and the International Human Genome Sequencing Consortium announced completion of the task on April 15, 2003 with an estimated accuracy of one error in 100,000 bases (15). The genome consists of 3 gigabases (three billion bases) of DNA sequence that code for approximately 30,000 genes, far fewer than was originally expected. However, it appears that this number of genes encodes 100,000 proteins through the process of alternative splicing whereby a gene’s exons or coding regions are spliced together in different ways to produce variant mRNA molecules that are translated into different proteins or isoforms of the same protein. Thus, the vast amounts of genomic data generated by the Human Genome Project have laid the foundation for an “-omic” revolution that includes, but is not limited to, the transcriptome, the set of expressed genes from a genome (16,17), the proteome, the set of proteins encoded by the genome (18), and the physiome, in which biochemical, biophysical, and anatomic information from cells, tissues, and organs will be integrated using computational methods to provide a model of the human body (19). Metabolomics and metabonomics are related terms that are sometimes used interchangeably in the literature. Metabolomics refers to the complete set of low–molecular-weight molecules (metabolites) present in a living system (cell, tissue, organ, or organism) at a particular developmental or pathological state. Metabonomics has been defined as the study of how the metabolic profile of biological systems changes in response to perturbations due to pathophysiologic stimuli, toxic exposures, dietary changes, among others (20,21). Pharmacometabonomics has been defined as “the prediction of the outcome, efficacy or toxicity, of a drug or xenobiotic intervention in an individual based on a mathematical model of preintervention metabolite signatures” (22), Chemogenomics is the application of combinatorial chemistry to generate libraries of small molecular weight compounds that can serve both as probes to investigate biological mechanisms and as lead compounds for drug development (23). Several resources that define these fields and their potential application to human health and disease are available on the Internet (Table 5.1).
Table 5.1 Internet Resources for Pharmacogenetics and Pharmacogenomics | |||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||
Basic Concepts and Definitions
Genetic variability results from gene mutation and the exchange of genetic information between chromosomes that occurs during meiosis. With the exception of sex-linked genes (genes occurring on the X or Y chromosomes), every individual carries two copies of each gene he or she
possesses. All copies of a specific gene present within a population may not have identical nucleotide sequences and these genetic polymorphisms contribute to the variability observed in that population. The presence of different nucleotides at a given position within a gene is called a single nucleotide polymorphism or “SNP” and SNPs are rapidly becoming an important component of the genomics lexicon. More recently, focus has shifted to characterizing haplotypes, collections of SNPs, and other allelic variations that are located close to each other and inherited together; creating a catalogue of haplotypes or “HapMap” is also a goal of the Human Genome Project (24). In genes where polymorphisms have been detected, alternative forms of the gene are called alleles. When the alleles at a particular gene locus are identical, a homozygous state exists whereas the term “heterozygous” refers to the situation in which different alleles are present at the same gene locus. The term genotype refers to an individual’s genetic constitution while the observable characteristics or physical manifestations constitute the phenotype, which is the net consequence of genetic and environmental effects.
possesses. All copies of a specific gene present within a population may not have identical nucleotide sequences and these genetic polymorphisms contribute to the variability observed in that population. The presence of different nucleotides at a given position within a gene is called a single nucleotide polymorphism or “SNP” and SNPs are rapidly becoming an important component of the genomics lexicon. More recently, focus has shifted to characterizing haplotypes, collections of SNPs, and other allelic variations that are located close to each other and inherited together; creating a catalogue of haplotypes or “HapMap” is also a goal of the Human Genome Project (24). In genes where polymorphisms have been detected, alternative forms of the gene are called alleles. When the alleles at a particular gene locus are identical, a homozygous state exists whereas the term “heterozygous” refers to the situation in which different alleles are present at the same gene locus. The term genotype refers to an individual’s genetic constitution while the observable characteristics or physical manifestations constitute the phenotype, which is the net consequence of genetic and environmental effects.
Human genetic variation can take many forms but is broadly divided into two general classes of variation: single nucleotide variations and structural variations. SNPs are the most prevalent class of genetic variation, and it has been estimated that there are approximately 11 million SNPs in the human genome. Structural variations encompass all differences in DNA sequence that involve more than one nucleotide. Insertions–deletions variants (indels) occur when a contiguous set of one or more nucleotides is absent in some individuals and present in others. Block substitutions involve variation of a string of contiguous nucleotides (or “block”) that differs between two genomes. Sequence inversions occur when the order of an entire block of nucleotides is reversed in a specific region of the genome. Finally, copy number variations (CNVs) refer to the deletion or duplication of identical or near-identical DNA sequences that may be thousands to millions of bases in size. Although they occur less frequently than SNPs, structural variations may constitute 0.5% to 1% of an individual’s genome and thus are the subject of intensive investigation for their contribution to phenotypic variation (25,26).
Pharmacogenetics, the study of the role of genetic factors in drug disposition, response, and toxicity, essentially relates allelic variation in human genes to variability in drug responses at the level of the individual patient. In other words, the promise of pharmacogenetics is to identify the right drug for the right patient. The field of pharmacogenetics classically has focused on the phenotypic consequences of allelic variation in single genes but often in the past, there was confusion between genotypic and phenotypic definitions of “polymorphism,” and thus a need to clarify the relationship between genetic concepts and the clinical relevance of a given phenotype. In 1991, Meyer proposed that pharmacogenetic polymorphism be defined as a monogenic trait caused by the presence in the same population of more than one allele at the same locus and more than one phenotype in regard to drug interaction with the organism. The frequency of the least common allele should be at least 1% (27). According to this definition, the key elements of pharmacogenetic polymorphisms are heritability, the involvement of a single gene locus, and the fact that distinct phenotypes are observed within the population only after drug challenge.
The vast majority of our current understanding of pharmacogenetic polymorphisms involves enzymes responsible for drug biotransformation. Clinically, individuals are classified as being “fast,” “rapid” or “extensive” metabolizers at one end of the spectrum, and “slow” or “poor” metabolizers at the other end of a continuum that may, depending on the particular enzyme, also include an intermediate metabolizer group. Pediatric pharmacogenetics involves an added measure of complexity since fetuses and newborns may be phenotypically “slow” or “poor” metabolizers for certain drug-metabolizing pathways, acquiring a phenotype consistent with their genotype at some point later in the developmental process as those pathways mature [e.g., glucuronidation, some cytochrome P450 activities (28,29,30)].
Although some authors use the terms “pharmacogenetics” and “pharmacogenomics” interchangeably, the latter term represents the marriage of pharmacology with genomics and is therefore considerably broader in scope. Pharmacogenomics can be defined as the study of the genome-wide response to small molecular weight compounds administered with therapeutic intent—finding the right drug for the right disease. Proteomics represents the systematic investigation of qualitative and quantitative changes in protein expression in a cell or tissue in response to disease or disease treatment. In this context, pharmacoproteomics involves characterizing the response of the proteome to therapeutic agents. Similarly, toxicogenomics and toxicoproteomics investigate the analogous response to environmental contaminants and other toxicants (31,32). In contrast to the focus of “pharmacogenetics” on single gene events, “pharmacogenomics” involves understanding how interacting systems or networks of genes influence drug responses (33). This definition is particularly appealing to pediatric health and disease since the concept of many genes acting in concert captures the essence of the developmental processes that characterize maturation from the time of birth through adulthood while retaining a focus on the individual.
It is safe to say that application of pharmacogenomic principles to pediatric medicine has received far less attention than its application to diseases affecting adults, and the scope of the field remains to be completely defined. However, developmental and pediatric pharmacogenomics necessarily must take into consideration the dynamic changes in gene expression that accompany maturation from embryonic life through fetal development, the neonatal period, infancy, childhood, and adolescence, for example, during organogenesis, as receptor systems and neural networks become established, and functional drug biotransformation capacity is acquired, among others. In other words, patterns of gene expression and the nature of the gene interactions that contribute to the pathogenesis of pediatric diseases (and thereby serve as potential targets for pharmacologic intervention) may only be discernable or relevant at specific critical points in the developmental continuum. Furthermore, variability in drug disposition (i.e., pharmacokinetics) and action (i.e., pharmacodynamics) that
ultimately impact drug response in pediatric patients can also be expected to change as children grow and develop. Finally, developmental and pediatric pharmacogenomic investigations can be distinguished from similar studies conducted in adults by the fact that drug or toxicant exposure at critical points in development may disrupt or alter the normal patterns of development—a genome-wide response to drug/toxicant exposure. This may have immediate, observable consequences, for example, fetal demise or major structural abnormalities such as those associated with retinoids (34) or other human teratogens. Of equal concern, however, is the possibility that drug exposure, or lack of effective drug treatment (35), may have unintended consequences on cognitive or behavioral development that do not manifest until much later in maturation. The remainder of this chapter will highlight examples of how pharmacogenomic approaches are currently improving pediatric pharmacotherapy and present several opportunities for future application.
ultimately impact drug response in pediatric patients can also be expected to change as children grow and develop. Finally, developmental and pediatric pharmacogenomic investigations can be distinguished from similar studies conducted in adults by the fact that drug or toxicant exposure at critical points in development may disrupt or alter the normal patterns of development—a genome-wide response to drug/toxicant exposure. This may have immediate, observable consequences, for example, fetal demise or major structural abnormalities such as those associated with retinoids (34) or other human teratogens. Of equal concern, however, is the possibility that drug exposure, or lack of effective drug treatment (35), may have unintended consequences on cognitive or behavioral development that do not manifest until much later in maturation. The remainder of this chapter will highlight examples of how pharmacogenomic approaches are currently improving pediatric pharmacotherapy and present several opportunities for future application.
Pharmacogenetic, Pharmacogenomic, Pharmacoproteomic, and Metabolomic Tools
Completion of the Human Genome Project was facilitated by several technological advances; the demands of genomic, proteomic, pharmacogenetic, and pharmacogenomic analyses have driven the development of an industry dedicated to the discovery and refinement of technologies capable of generating large datasets of information derived from DNA, RNA, proteins, and small endogenous molecules. These tools are used widely to investigate disease pathogenesis but are equally applicable to investigations of variability in drug disposition and response.
Pharmacogenetic Tools
Historically, pharmacogenetic analyses have been dependent upon phenotyping studies to estimate enzyme activity in vivo at a specific time point as well as genotyping strategies to identify and characterize SNPs and other forms of genetic variation. Phenotyping studies are best conducted with a probe compound carefully selected to ensure that its biotransformation is primarily dependent upon a single target enzyme and varies quantitatively with the level of protein expression (i.e., phenotyping data correlate with the level of enzyme activity in vitro, the fractional clearance of the probe and other substrates of the target enzyme in vivo, and biotransformation of the probe is increased or decreased in the presence of inducers and inhibitors, respectively) (36). An ideal probe should involve noninvasive sampling strategies, such as collection or urine or expired air rather than blood samples, especially when phenotyping studies are to be conducted in children. Finally, candidate phenotyping probes should be widely available (nonprescription status, preferably) and have a wide margin of safety. For pediatric studies, the phenotyping probe should be selected from compounds that are likely to be administered to children and perceived as safe by parents, caregivers, and ethics committees (e.g., dextromethorphan as opposed to debrisoquine or sparteine for CYP2D6). The advantages and disadvantages of phenotyping probes commonly utilized in adult studies have been comprehensively and critically evaluated by Streetman et al. (37). Dextromethorphan and caffeine are commonly used in pediatric phenotyping studies with nontherapeutic intent (38,39,40,41). However, other accepted phenotyping probes, such as midazolam for CYP3A4 and omeprazole for CYP2C19, may be utilized in selected patient populations where their use is required for therapeutic purposes.
The genotyping component of pharmacogenetic studies has undergone tremendous change over the past 30 years. Historically, studies were conducted at the level of individual genes using rather insensitive DNA hybridization techniques (42) to detect differences in the patterns of DNA fragments generated following digestion of genomic DNA with restriction endonucleases (enzymes that cleave DNA molecules at specific nucleotide sequences). The restriction fragment length polymorphism (RFLP) technique was later coupled with the polymerase chain reaction (PCR–RFLP) to allow a specific region surrounding the SNP of interest to be amplified from small amounts of genomic DNA followed by endonuclease digestion to identify the allelic variant(s) present (43). PCR–RFLP techniques have been widely used to study cytochrome P450 polymorphisms (44,45,46), among others, but they are too labor-intensive for routine use in genomic applications, such as fine-mapping of disease loci or candidate gene association studies, which involve the analysis of multiple SNPs in thousands of genes. Many innovative genotyping technologies continue to be developed, and some have received approval by the Food and Drug Administration for use in clinical settings. The Roche Amplichip CYP450 Test was the first such device to receive FDA approval (December 17, 2004 and January 10, 2005), followed by products from Third Wave Technologies, Inc. (August 18, 2005), Verigene (September 17, 2007), Autogenomics, Inc. (January 23, 2008), ParagonDx, LLC (April 28, 2008), Osmetech Molecular Diagnostics (July 17, 2008), and Trimgen Corporation (February 6, 2009). In general, applications are limited to one or two genes, such as CYP2C9 and VKORC1 genotyping to guide warfarin therapy or genotyping of UGT1A1 to reduce the risk of irinotecan toxicity.
Pharmacogenomic Tools
In contrast to pharmacogenetic studies that typically target single genes, pharmacogenomic analyses are considerably broader in scope since they focus on complex and highly variable drug-related phenotypes (e.g., valproic acid hepatotoxicity or weight gain, tumor response to cancer chemotherapy, drug response in asthma, epilepsy, attention deficit and hyperactivity disorders). Comparison of gene expression profiles in cells from treatment-responsive and nonresponsive patients is a readily accessible pharmacogenomic phenotype. These types of studies utilize microarray or “gene-chip” technology to monitor global changes in expression of thousands of genes simultaneously—“global gene
profiling.” In essence, a microarray consists of a matrix of DNA fragments (probes) precisely positioned (i.e., coordinates are known) at high density on a solid support, such as a glass slide or a filter. The probes serve as molecular detectors for mRNA in the sample. Common experimental designs involve labeling mRNA (or cDNA) from a control sample with one fluorescent dye and mRNA (or cDNA) from the disease/treatment sample with a second fluorescent dye, using an experimental strategy that allows expression to be compared between the sample pair. Alternatively, samples from many patients can be run, each on a single chip, and the results from all the chips subjected to normalization procedures that allow the gene expression patterns from treated patients and controls, or responders and nonresponders, to be compared. Thus, the expression pattern of thousands of genes can be analyzed in a single sample with the underlying hypothesis that the measured intensity for each arrayed gene represents its relative expression level. Because of the massive amounts of data generated in these experiments, sophisticated computational methods and data-mining tools are necessary to reveal patterns of expression that distinguish between the populations under investigation. Potential applications of gene expression profiling include improved disease classification and risk stratification in oncology; pathogen detection, subtyping, and characterization of antibiotic resistance; neonatal screening for genetic disorders and prediction of drug response and adverse drug reactions (47). As an example, this approach has been widely used to address treatment resistance in acute lymphoblastic leukemia and has provided new insights into the mechanistic basis of drug resistance and the genomic basis of interindividual variability in drug response (48,49).
profiling.” In essence, a microarray consists of a matrix of DNA fragments (probes) precisely positioned (i.e., coordinates are known) at high density on a solid support, such as a glass slide or a filter. The probes serve as molecular detectors for mRNA in the sample. Common experimental designs involve labeling mRNA (or cDNA) from a control sample with one fluorescent dye and mRNA (or cDNA) from the disease/treatment sample with a second fluorescent dye, using an experimental strategy that allows expression to be compared between the sample pair. Alternatively, samples from many patients can be run, each on a single chip, and the results from all the chips subjected to normalization procedures that allow the gene expression patterns from treated patients and controls, or responders and nonresponders, to be compared. Thus, the expression pattern of thousands of genes can be analyzed in a single sample with the underlying hypothesis that the measured intensity for each arrayed gene represents its relative expression level. Because of the massive amounts of data generated in these experiments, sophisticated computational methods and data-mining tools are necessary to reveal patterns of expression that distinguish between the populations under investigation. Potential applications of gene expression profiling include improved disease classification and risk stratification in oncology; pathogen detection, subtyping, and characterization of antibiotic resistance; neonatal screening for genetic disorders and prediction of drug response and adverse drug reactions (47). As an example, this approach has been widely used to address treatment resistance in acute lymphoblastic leukemia and has provided new insights into the mechanistic basis of drug resistance and the genomic basis of interindividual variability in drug response (48,49).
Whole genome genotyping technologies now make it possible to interrogate genetic variation more than a million sites throughout an individual genome for SNP and CNV analyses using a single “chip.” The majority of genome-wide association (GWA) studies are conducted with “SNP chips” utilizing one of two commercial platforms, and the approach recently has been applied to several pediatric diseases. A study of Kawasaki disease identified a set of functionally related genes potentially related to inflammation, apoptosis, and cardiovascular pathology (50). The results of the study provide novel insights into the pathogenesis of the disorder and lead to the possibility of identifying new targets for therapeutic intervention. Similarly, GWA studies in patients with early onset asthma (51) and pediatric inflammatory bowel disease (52) have been implemented as a new strategy to identify novel genes in disease pathogenesis. Genome-wide association studies (GWAS) are also being applied to identify genetic associations with drug dosing, response and efficacy, as reported for warfarin (53) and clopidogrel (54), and risk for drug-induced toxicity, as has been described for statin-induced myopathy (55). One of the features of GWAS that is becoming more prevalent is the use of Manhattan plots. An example is presented in Figure 5.2.
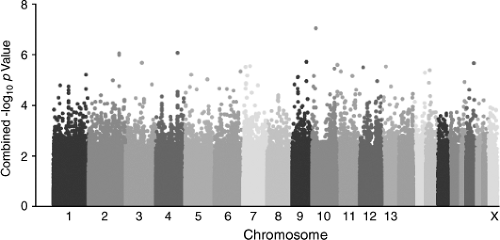 Figure 5.2. Example of a Manhattan plot from a genome-wide association study. This type of plot gains its name from the similarity of such a plot to the Manhattan skyline and presents the genome-wide significance of several hundred thousand SNPs distributed throughout the genome with the trait or phenotype of interest. Along the abscissa, each SNP is plotted according to its chromosomal coordinate, with each color representing an individual chromosome from chromosome 1 to the X chromosome. The ordinate axis represents the inverse log10 of the p value for the association. SNPs exceeding a particular threshold are subject to further verification and validation. (Originally published as Figure 2 in Yang JJ et al., Genome-wide interrogation of germline genetic variation associated with treatment response in childhood acute lymphoblastic leukemia. JAMA 2009;301: 393–403 (164). Reprinted with permission.) |
The most recent addition to the genomic toolbox is next-generation sequencing technology (56). The major difference between next-generation sequencing methods and the older, more established Sanger capillary sequencing method is the vast amount of sequencing data that can be generated by the next-generation technologies, which utilize massively parallel and short read strategies to sequence DNA and RNA templates. Whereas it has been estimated that 500 days of runtime were required to generate one gigabase (one billion nucleotides) of data, the newer next-generation sequencers can produce this same amount of data in half a day at 1/100th of the cost. However, the short 50 to 75 bp-read lengths provide computational challenges related to assembly of the short sequence reads to produce an accurate contiguous genomic
sequence. Application of next-generation sequencing to RNA templates referred to as RNA-Seq allows deeper coverage and higher resolution of RNA sequence compared to microarray technology and has revolutionized transcriptomics. RNA-Seq has provided new insights into gene regulation, alternative splicing, and allelic variation of transcripts increasing dramatically our understanding of the diversity of protein products encoded by the genome (57,58).
sequence. Application of next-generation sequencing to RNA templates referred to as RNA-Seq allows deeper coverage and higher resolution of RNA sequence compared to microarray technology and has revolutionized transcriptomics. RNA-Seq has provided new insights into gene regulation, alternative splicing, and allelic variation of transcripts increasing dramatically our understanding of the diversity of protein products encoded by the genome (57,58).
Pharmacoproteomic Tools
Proteomic studies face limitations that are not experienced in genomic or transcriptomic applications. For example, no amplification method analogous to PCR exists to increase the quantity of protein from small amounts of starting material, and the final protein product in a sample may be the result of alternative splicing events and several posttranslational modifications. Therefore, many different techniques are required to detect, quantify and identify proteins in a sample (expression proteomics) and to characterize protein function in terms of activity and protein–protein or protein–nucleic acid interactions (functional proteomics) (59). At present, two-dimensional electrophoresis (2DE) coupled with mass spectral detection (2DE-MS) represents the mainstay of expression proteomics. Matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) mass spectrometry is a commonly used approach, and data generated are compared with theoretically derived peptide mass databases for protein identification. Proteomic analyses of drug-induced toxicities of potential relevance to pediatric pharmacotherapy have identified candidate protein targets of acetaminophen hepatotoxicity (60) and gentamicin nephrotoxicity (61). More recently, application of surface-enhanced laser desorption/ionization (SELDI) based approaches has revealed the potential of proteomic technology to identify biomarkers of response to drug treatment in pediatric diseases, such as juvenile idiopathic arthritis (62) and the response of pediatric nephritic syndrome to steroids (63).
Metabolomic Tools
Several analytical approaches can be utilized for metabolomic and metabonomic investigations, depending on the analytes of interest, but the most used platforms are NMR spectroscopy and liquid or gas chromatography coupled with tandem mass spectral detection (64). One strategy for metabolomic studies is to use a global profiling approach to measure the concentrations of all small molecules present in a sample or changing in response to a perturbation. Subsequent application of pattern recognition algorithms defines a metabolic phenotype that can be associated with a particular end point, such as disease state, drug response, or drug toxicity. Metabolomics can be integrated with other genomic technologies. Combining metabolomic and genome-wide genotyping data has revealed common genetic variations that are associated with variability in metabolic phenotypes involving the corresponding biochemical pathways (65). Metabolomics in conjunction with simultaneous gene expression analysis of the transcriptome provides additional mechanistic insights leading to a more “systems-based” understanding of cellular processes, especially in the context of dug-related perturbations (66). Relatively few metabolomic studies have been conducted in children, but NMR has been used to investigate age-related changes in the metabolome between newborns and children 12 years of age (67).
Applications of Pediatric Pharmacogenetics and Pharmacogenomics
Drug Biotransformation and Concentration-Dependent Toxicity
Clinical observation of patients with high drug concentrations/excessive or prolonged drug responses, together with the realization that the biochemical traits (subsequently identified as proteins involved in drug biotransformation) were inherited, provided the origins of the concept of pharmacogenetics. Indeed, with few exceptions, the major consequence of pharmacogenetic polymorphisms in drug-metabolizing enzymes is concentration-dependent toxicity due to impaired drug clearance and, to a lesser extent, reduced conversion of prodrugs to therapeutically active compounds. For most cytochromes P450, genotype–phenotype relationships are influenced by development in that fetal expression is limited (with the exception of CYP3A7) and functional activity is acquired postnatally in isoform-specific patterns. Furthermore, clearance of some compounds appears to be greater in children relative to adults, obscuring the correlation between genotype and phenotype in neonatal life through adolescence (28). The ontogeny of several phase I and phase II drug biotransformation pathways has been exhaustively reviewed (68), and comprehensive reviews of cytochromes P450 pharmacogenetics in general (69) and for individual drug biotransformation enzymes, such as CYP2B6 (70), CYP2C9 (71), CYP2C19 (72), CYP2D6 (73,74), the CYP3A subfamily (75), uridine diphospho-glucuronosyl transferases or UGTs (76,77), sulfotransferases or SULTs (78), N-acetyltransferases or NATs (79), and thiopurine S-methyltransferase or TPMT (80) are also available. Salient features of the more common polymorphisms of clinically relevant drug-metabolizing enzymes are now discussed briefly.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


