Chapter 56 Pediatric Pharmacogenetics, Pharmacogenomics, and Pharmacoproteomics
Interindividual variability in the response to similar doses of a given medication is an inherent characteristic of adult and pediatric populations. The role of genetic factors in drug disposition and response, pharmacogenetics, has resulted in many examples of how variations in human genes can lead to interindividual differences in pharmacokinetics and drug response at the level of individual patients. Just as in adults, pharmacogenetic variability contributes to the broad range of drug responses observed in children at any given age or developmental stage. Therefore, it is expected that children will benefit from the promise of personalized medicine: identifying the right drug for the right patient at the right time (see Fig. 56-1 on the Nelson Textbook of Pediatrics website at  www.expertconsult.com). However, pediatricians are keenly aware that children are not merely small adults. Numerous maturational processes occur from birth through adolescence, and using information resulting from the Human Gene Project and related initiatives must take into account the changing patterns of gene expression that occur over development to improve pharmacotherapeutics in children.
www.expertconsult.com). However, pediatricians are keenly aware that children are not merely small adults. Numerous maturational processes occur from birth through adolescence, and using information resulting from the Human Gene Project and related initiatives must take into account the changing patterns of gene expression that occur over development to improve pharmacotherapeutics in children.
Pharmacogenetics, Pharmacogenomics, and the Concept of Personalized Medicine
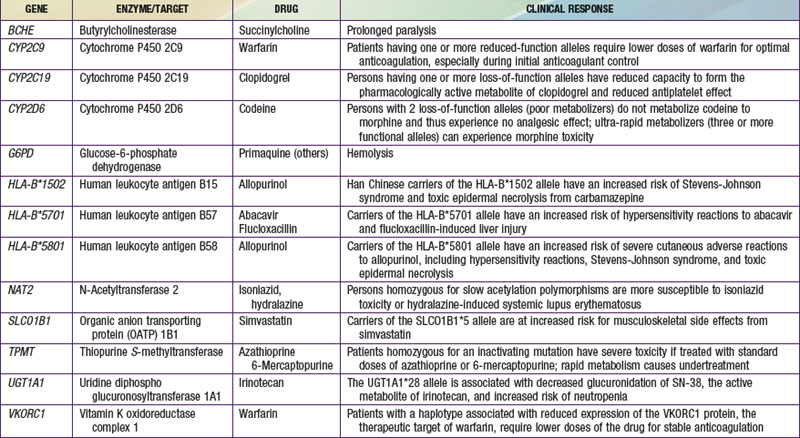
The terms pharmacogenomics and pharmacogenetics tend to be used interchangeably, and precise consensus definitions are often difficult to determine. Pharmacogenetics classically is defined as the study or clinical testing of genetic variations that give rise to interindividual response to drugs. The earliest examples of pharmacogenetic traits include specific adverse drug reactions, such as unusually prolonged respiratory muscle paralysis caused by succinylcholine, hemolysis associated with antimalarial therapy, and isoniazid-induced neurotoxicity, all of which were found to be a consequence of inherited variations in enzyme activity. The importance of pharmacogenetic differences is exemplified by the fact that the half-lives of several drugs are more similar in monozygotic twins than in dizygotic twins. However, in addition to pharmacogenetic differences, environmental factors (diet, smoking status, concomitant drug or toxin exposure), physiologic variables (age, sex, disease, pregnancy), and patients’ compliance all contribute to variations in drug metabolism and response. Ethnicity is another potential genetic determinant of drug variability. Chinese patients who are HLA-B*1502 positive have an increased risk of carbamazepine-induced Stevens-Johnson syndrome; white patients who are HLA-B*5701 positive have an increased risk of hypersensitivity to abacavir (Table 56-1).
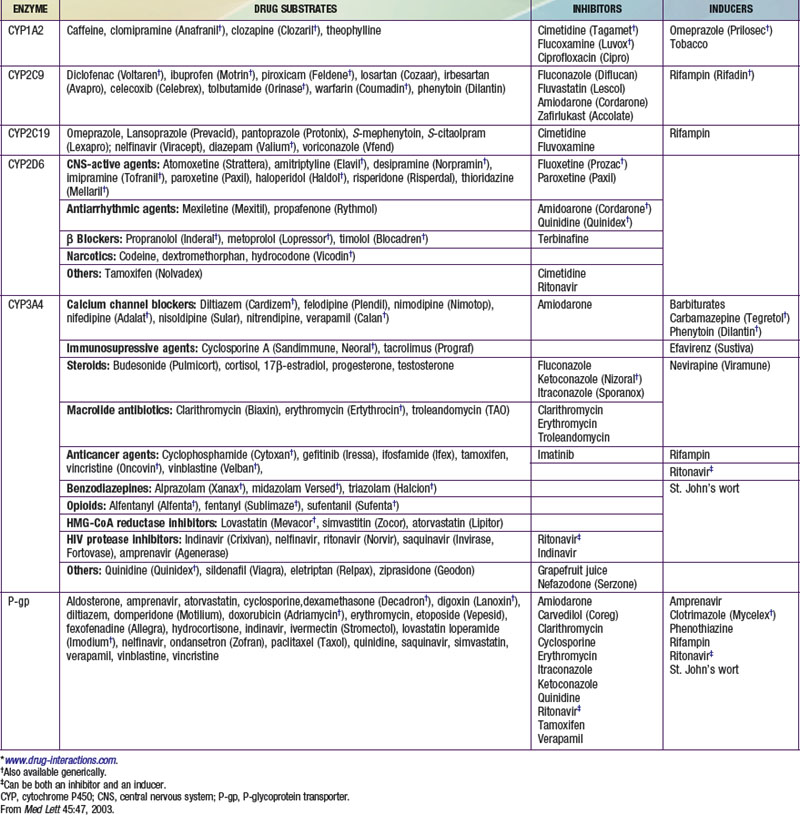
Pharmacokinetics describes what the body does to a drug. It is often studied in conjunction with pharmacodynamics, which explores what a drug does to the body. The pharmacokinetic properties of a drug are determined by the genes that control the drug’s disposition in the body (absorption, distribution, metabolism, excretion [ADME]). Drug-metabolizing enzymes and drug transporters play a particularly important role in this process (Table 56-2), and the functional consequences of genetic variations in many drug-metabolizing enzymes have been described among subjects of similar and different ethnic groups. The most common clinical manifestation of pharmacogenetic variability in drug biotransformation is an increased risk of concentration-dependent toxicity resulting from reduced clearance and consequent drug accumulation. An equally important manifestation of this variability is lack of efficacy resulting from variations in metabolism of prodrugs. The pharmacogenetics of drug receptors and other target proteins involved in signal transduction or disease pathogenesis can also be expected to contribute significantly to interindividual variability in drug disposition and response.
Table 56-2 SOME IMPORTANT RELATIONSHIPS BETWEEN DRUGS AND CYTOCHROME P450 ENZYMES* AND P-GLYCOPROTEIN TRANSPORTER
Definition of Pharmacogenetics Terms
Genetic polymorphisms (variations) result when copies of a specific gene in a population do not have identical nucleotide sequences. The term allele refers to one of a series of alternative DNA sequences for a particular gene. In humans, there are two copies of every gene. An individual’s genotype for a given gene is determined by the set of alleles that the individual possesses. The most common form of genetic variation involves a single base change at a given location, referred to as a single-nucleotide polymorphism (SNP) (Chapters 72 and 74).
The term genotype refers to an individual’s genetic constitution, whereas the observable characteristics or physical manifestations constitute the phenotype, which is the net consequence of genetic and environmental effects (Chapters 72–77). Pharmacogenetics focuses on the phenotypic consequences of allelic variation in single genes.
Developmental or Pediatric Pharmacogenetics and Pharmacogenomics
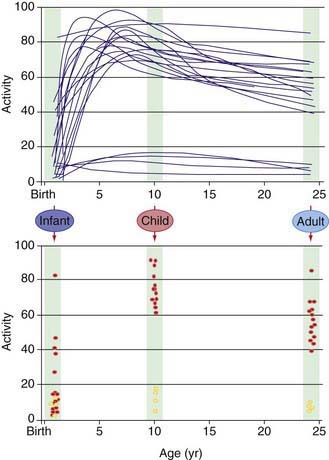
Our understanding of pharmacogenetic principles involves enzymes responsible for drug biotransformation. Individuals are classified as being “fast,” “rapid,” or “extensive” metabolizers at one end of the spectrum, and “slow” or “poor” metabolizers at the other end. This might or might not also include an “intermediate” metabolizer group, depending on the particular enzyme. With regard to biotransformation, children are more complex than adults because fetuses and newborns may be phenotypically slow or poor metabolizers for certain drug-metabolizing pathways because of their stage of development, and they can acquire a phenotype consistent with their genotype at some point later in the developmental process as they mature. Examples of drug-metabolizing pathways that are significantly affected by ontogeny include glucuronidation and some activities of the cytochrome P450 isoenzymes (CYPs) (Chapter 57). It is also apparent that not all infants acquire drug metabolism activity at the same rate. This is due to interactions between genetics and environmental factors. Interindividual variability in the trajectory (i.e., rate and extent) of acquired drug biotransformation capacity may be considered a developmental phenotype (Fig. 56-2), and it helps to explain the considerable variability in some CYP activities observed immediately after birth.
Pharmacogenetic, Pharmacogenomic, Pharmacoproteomic, and Metabolomic Tools
Several genotyping platforms are approved by the U.S. Food and Drug Administration (FDA) and are entering the clinical arena. The Roche Amplichip CYP450 Test was the first such device to receive FDA approval, and at least 6 additional products have since received approval. In general, applications are limited to 1 or 2 genes, such as CYP2C9 and VKORC1 genotyping to guide warfarin therapy or genotyping of UGT1A1 to reduce the risk of irinotecan toxicity. A more comprehensive chip that covers >90% of the ADME markers as defined by the PharmaADME group (http://pharmaadme.org) is available for drug development and research purposes.
In contrast to pharmacogenetic studies that typically target single genes, pharmacogenomic analyses are considerably broader in scope and focus on complex and highly variable drug-related phenotypes, with targeting of many genes. Genome-wide genotyping technologies make it possible to evaluate genetic variation at more than a million sites throughout an individual genome for SNP and CNV analyses using a single chip. One goal of this type of study is to identify novel genes involved in disease pathogenesis that can lead to new therapeutic targets. Genome-wide association studies (GWAS) are also being applied to identify genetic associations with response to drugs, such as warfarin and clopidogrel, and risk for drug-induced toxicity, including statin-induced myopathy and flucloxacillin hepatotoxicity. The Manhattan plot, a form of data presentation for GWAS, is one way to represent these data (Fig. 56-3A).
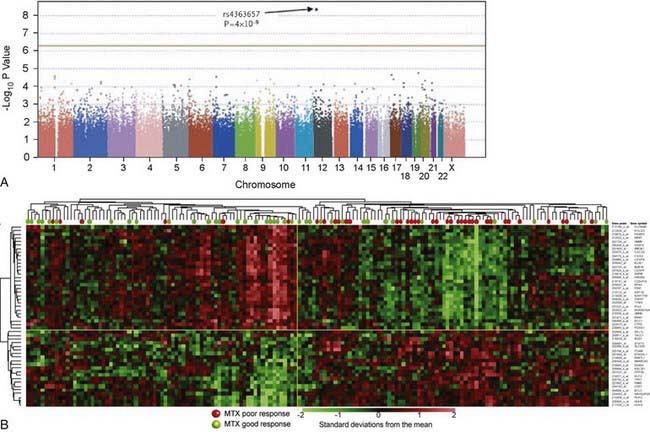
Investigating differential gene expression before and after drug exposure has the potential to correlate gene expression with variable drug responses and possibly uncover the mechanisms of tissue-specific drug toxicities. These types of studies use microarray technology to monitor global changes in expression of thousands of genes (the transcriptome) simultaneously. The underlying hypothesis of these global gene-profiling studies is that the measured intensity for each arrayed gene represents its relative expression level. Gene expression profiling data are used to improve disease classification and risk stratification and are used commonly in oncology. For example, this approach has been widely used to address treatment resistance in acute lymphoblastic leukemia and has provided clinically relevant insights into the mechanistic basis of drug resistance and the genomic basis of interindividual variability in drug response. Subsets of transcripts, or gene expression signatures, are being investigated as potential prognostic indicators for identifying patients at risk for treatment failure (Fig. 56-3B).
Developmental Pharmacogenetics of Drug Biotransformation: Applications to Pediatric Drug Therapy Practice
The major consequence of pharmacogenetic polymorphisms in drug-metabolizing enzymes is concentration-dependent toxicity resulting from impaired drug clearance. In certain cases, reduced conversion of prodrug to therapeutically active compounds is also of clinical importance (see Table 56-2). Chemical modification of drugs via biotransformation reactions generally results in termination of biologic activity through decreased affinity for receptors or other cellular targets as well as more rapid elimination from the body. The process of drug biotransformation can be very complex, but it is characterized by 3 important features. First is the concept of broad substrate specificity: A single isozyme can metabolize a large variety of chemically diverse compounds. Second, many different enzymes may be involved in the biotransformation of a single drug (enzyme multiplicity). Finally, a given drug can undergo several different types of reactions. One example of this product multiplicity occurs with racemic warfarin, where at least 7 different hydroxylated metabolites are produced by different CYP isoforms.
Drug biotransformation reactions are conveniently classified into 2 main types, phase I and phase II reactions, which occur sequentially and serve to terminate biologic activity and enhance elimination (Chapter 57). Phase I reactions introduce or reveal (via oxidation, reduction, or hydrolysis) a functional group within the substrate drug molecule that serves as a site for a phase II conjugation reaction. Phase II reactions involve conjugation with endogenous substrates, such as acetate, glucuronic acid, glutathione, glycine, and sulfate. These reactions further increase the polarity of an intermediate metabolite, make the compound more water soluble, and thereby enhance its renal excretion. Interindividual variability in drug biotransformation activity (for both phase I and phase II reactions) is a consequence of the complex interplay among genetic (genotype, sex, race, or ethnic background) and environmental (diet, disease, concurrent medication, other xenobiotic exposure) factors. The pathway and rate of a given compound’s biotransformation is a function of each individual’s unique phenotype with respect to the forms and amounts of drug-metabolizing enzymes expressed.
The CYPs are quantitatively the most important of the phase I enzymes. These heme-containing proteins catalyze the metabolism of many lipophilic endogenous substances (steroids, fatty acids, fat-soluble vitamins, prostaglandins, leukotrienes, and thromboxanes) as well as exogenous compounds, including a multitude of drugs and environmental toxins. CYP nomenclature is based on evolutionary considerations. CYPs that share at least 40% homology are grouped into families denoted by an Arabic numeral after the CYP root. Subfamilies, designated by a letter, appear to represent clusters of highly related genes. Members of the human CYP2 family, for example, have >67% amino acid sequence homology. Individual CYPs in a subfamily are numbered sequentially (e.g., CYP3A4, CYP3A5). CYPs that have been identified as being important in human drug metabolism are predominantly found in the CYP1, CYP2, and CYP3 gene families. Importantly, enzyme activity may be induced or inhibited by various agents (see Table 56-2).
CYP2D6
The CYP2D6 gene locus is highly polymorphic, with >75 allelic variants identified to date (http://www.imm.ki.se/CYPalleles/cyp2d6.htm; see Table 56-2). Individual alleles are designated by the gene name (CYP2D6) followed by an asterisk, and an Arabic number. By convention, CYP2D6*1
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


