Pediatric Ophthalmology
Elias I. Traboulsi
NORMAL DEVELOPMENT OF THE EYE
Ocular and orbital structures are derived from two populations of cells: mesodermal and ectodermal. The vascular endothelium, extraocular muscles, and part of the temporal sclera are derived from mesoderm. The three types of ectodermal cells that contribute to the remaining ocular structures are neural ectoderm, neural crest cells, and surface ectoderm. Most mesenchymal tissues of the eye and orbit are derived from the neural crest.
At the end of the second week of gestation, through adjacent mesodermal induction, the ocular primordia arise from the neural plate. The optic vesicles form from neuroectoderm and approach surface ectoderm to induce the lens placode. At 4 weeks’ gestation, the optic vesicle and lens placode invaginate, and vessels of the hyaloid system are incorporated into the globe through this formed embryonic fissure, located inferiorly in the developing globe. At the beginning of the fifth week, the embryonic fissure closes, and by the sixth week, the entire double-layered optic cup is formed. The inner neuroectodermal cell layer becomes the multilayered sensory retina, and the outer layer becomes the retinal pigment epithelium. The neuroectodermal layers induce the surrounding mesenchyme to produce the stroma of the choroid and the melanocytes of the uveal tract. The collagenous coats of the eye, the bones, and soft tissues of the orbit and the sheaths of the optic nerve are derived from the neural crest. A number of developmental genes, the best studied of which being the PAX6 gene, play an essential role in normal ocular development. These genes are expressed in the embryonic eye structures and are mutated in some ocular malformations such as aniridia.
Normal Milestones
A normal pupillary response indicates functioning afferent and efferent visual pathways. This response is usually present by 31 weeks’ gestation. The blink response to light occurs by 30 weeks’ gestation. A blink response to threat is not observed until 5 months of age. Estimates of visual acuity in young infants are based on the ability to fixate and follow targets, on the presence or absence of nystagmus, and on the indifference to occlusion of either eye. The infant would fuss if a seeing eye is closed and a poorly seeing eye is left uncovered. In older infants, good vision allows normal play behavior and interaction with parents and provides the children with the ability to notice and find small objects and distant visual targets. Human faces are seen by infants as young as a few hours of age. Questioning a mother about her child’s response to her face and smile is a useful clue to the child’s ability to see in the absence of major neurologic and developmental impairment. The presence of an optokinetic response to a rotating drum with vertical or horizontal stripes is also used to rule out blindness in infants with suspected total visual loss (vide infra).
Acuity Measurement in Infancy
Objective techniques of visual acuity assessment in infancy include optokinetic nystagmus (OKN), visual-evoked potentials (VEPs), and forced-choice preferential looking (FCPL) techniques. In the OKN technique, a drum with black and white stripes is rotated so that the stripes are moved in an arc across 180° of the infant’s visual field. This results in an involuntary horizontal jerk nystagmus in the seeing infant. The fast phase of the nystagmus is in the direction opposite that of the moving stripes. OKN response to a smaller stripe width indicates better visual acuity.
Analysis of the latency and amplitude of VEPs elicited by phase-alternated checkerboards of variable size or square-wave gratings can also be used to determine visual acuity in infants. The level of acuity is directly proportional to the amplitude of the VEP and inversely proportional to the latency of the evoked response. VEPs are not affected by acoustic stimuli, movements of the observer’s limbs, or some eye movements.
FCPL techniques are based on the observation that infants prefer to look at a patterned stimulus than on a field of homogeneous gray color of equal overall luminance. The Teller Acuity Cards II are probably the most frequently used and reliable instruments that rely on this technique. In this examination, the infant is presented simultaneously with a patterned stimulus consisting of black and white stripes on one side, and with a gray screen of space-average luminance equal to the patterned stimulus on the other side. The observer, who is peeping at the infant through a hole between the two stimuli, is masked to the relative position of the striped or checkered stimulus and the gray screen. The observer records the visual response of the child by noting which side the child’s eyes are pointed to as patterns with decreasing stripe widths are presented. Acuity is estimated from the smallest stripe width the child prefers over the homogeneous field. Although tedious, this is a useful behavioral technique for assessing visual acuity and diagnosing a variety of ocular diseases. Its most common application is the determination of the response amblyopia to treatment in the preverbal child.
Table 128.1 summarizes estimates of normal visual acuity during the first year of life based on the three methods described. Each of these methods has its limitations and inconsistencies, but all have proven useful in the objective assessment of visual acuity in infants. Occasionally, infants have a maturational delay in visual development in the absence of structural ocular abnormalities or nystagmus. There is frequently a concomitant delay in motor skill development. This condition should only be diagnosed if organic causes of reduced vision such as media opacities, optic atrophy, and retinal dystrophies such as Leber congenital amaurosis are excluded. An ocular examination and a normal or only slightly reduced
electroretinogram differentiate delayed visual maturation from the other conditions.
electroretinogram differentiate delayed visual maturation from the other conditions.
TABLE 128.1. DEVELOPMENT OF VISUAL ACUITY IN INFANCY AS ASSESSED BY VARIOUS TECHNIQUES | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Acuity Measurement in Older Children
Allen pictures or Lea symbols are most commonly used to assess vision in 2- to 3-year-old children. The pictures or symbols are either projected in decreasing sizes at a fixed distance or are presented to the child on cards of a fixed size at increasing distances. In the Sheridan-Gardiner test, the child is shown geometric shapes, letters, or patterns of decreasing sizes and asked to point to identical patterns on a chart held at reading distance. The E game can be used for the child who is older than 3 years of age but cannot yet read letters. The child is asked to point his or her fingers or hand in the direction of the open end of the horizontal lines of E’s of decreasing sizes on a chart or on cards. It may be necessary to teach the child to play the E game or to recognize Allen pictures at home before the examination.
Caution must be used in interpreting the visual acuity of children tested by a method that involves the recognition of letters. Children will not admit that they do not know the letters and can mislead the examiner. Children with unilateral visual loss may also “peak” from the better eye if it is not adequately occluded.
OPHTHALMOLOGIC EXAMINATION
The recommendations of the American Academy of Ophthalmology and the American Association for Pediatric Ophthalmology and Strabismus are that an ophthalmologic examination be performed whenever questions arise about the health of the visual system of a child of any age. The recommendations also include the following:
A pediatrician, family physician, nurse practitioner, or physician assistant should examine a newborn’s eyes for general eye health including a red reflex test in the nursery. An ophthalmologist should be asked to examine all high risk infants, that is, those at risk to develop retinopathy of prematurity (ROP); those with a family history of retinoblastoma, glaucoma, or cataracts in childhood; those with retinal dystrophy/degeneration or systemic diseases associated with eye problems; or when any opacity of the ocular media or nystagmus (purposeless rhythmic movement of the eyes) is seen. Infants with neurodevelopmental delay should also be examined by an ophthalmologist.
All infants by 6 months to 1 year of age should be screened for ocular health including a red reflex test by a properly trained health care provider such as an ophthalmologist, pediatrician, family physician, nurse, or physician assistant during routine well-baby follow-up visits.
Vision screening should also be performed between 3 and 3½ years of age. Vision and alignment should be assessed by a pediatrician, family practitioner, ophthalmologist, optometrist, orthoptist, or individual trained in vision assessment of preschool children. Emphasis should be placed on checking visual acuity as soon as a child is cooperative enough to complete the examination. Generally, this occurs between ages 2½ and 3½. It is essential that a formal testing of visual acuity be performed by the age of 5 years.
Some evidence currently exists to suggest that photoscreening may be a valuable adjunct to the traditional screening process, particularly in preliterate children.
Further screening examinations should be done at routine school checks or after the appearance of symptoms. Routine comprehensive professional eye examination of the normal asymptomatic child has no proven medical benefit.
School-aged children who pass standard vision screening tests but who demonstrate difficulties learning to read should be referred to reading specialists such as educational psychologists for evaluation for language processing disorders such as dyslexia. There is not adequate scientific evidence to suggest that “defective eye teaming” and “accommodative disorders” are common causes of educational impairment. Hence, routine screening for these conditions is not recommended.
Visual acuity can be assessed in the pediatrician’s office with Allen cards or Sheridan-Gardiner cards for children 3 to 5 years of age, and with Snellen acuity charts for older children who know the alphabet well. The E chart may also be used. In preverbal or retarded children, symmetry of visual acuity between the two eyes can be determined by the pattern of fixation. In the latter, vision is recorded as being central or eccentric, steady or interrupted by abnormal or involuntary movements, and maintained or preferred to one eye or the other.
The primary health care provider can check ocular alignment using the cover test, the cover-uncover test, or the Hirschberg corneal light reflex test. The light reflexes should be symmetrically located in the corneas. The cover and cover-uncover tests are based on the observation that children with strabismus use one eye for fixation while the other eye is deviated. When the fixating eye is covered, the deviated eye moves in or out to pick up fixation. If the eye moves from the nasal side to the temporal side, it is esotropic; if it moves from the temporal side toward the nose, it is exotropic. Pupillary responses are checked with a bright light source, and the direct (i.e., stimulated pupil constricts) and consensual (i.e., other pupil constricts when light is shined in one eye) light reflexes are recorded. An afferent pupillary defect during the swinging light test (i.e., pupil dilates instead of constricting as light is moved from the other eye back to the one with the dilating pupil) usually indicates an optic nerve problem such as atrophy or neuritis on the side of the afferent defect.
The direct ophthalmoscope can be used to check for the presence of cataracts or other ocular media opacities. A +10
diopter lens is dialed into the ophthalmoscope, and the ophthalmoscope light is shined into the pupil from a distance of about 1 m while the observer is looking into the ophthalmoscope; cataracts appear as black shadows over a red background. Direct ophthalmoscopy allows examination of the disc, macula, and blood vessels in the posterior pole area. Dimming the light of the ophthalmoscope and shining the light into the pupil from a distance of less than an inch while the child is looking at a distance with the other eye maximizes the chance of having a good view of the disc and posterior pole.
diopter lens is dialed into the ophthalmoscope, and the ophthalmoscope light is shined into the pupil from a distance of about 1 m while the observer is looking into the ophthalmoscope; cataracts appear as black shadows over a red background. Direct ophthalmoscopy allows examination of the disc, macula, and blood vessels in the posterior pole area. Dimming the light of the ophthalmoscope and shining the light into the pupil from a distance of less than an inch while the child is looking at a distance with the other eye maximizes the chance of having a good view of the disc and posterior pole.
Children with strabismus or those with evident or suspected ocular abnormalities should be immediately examined by an ophthalmologist, as should infants suspected to have impaired vision. Children with syndromes or diseases known to involve the eye, those with a family history of early onset of ocular disease, or those with developmental delay or suspicion of visual handicap should also be examined immediately. Visual handicaps need to be ruled out in children with scholastic failure or learning disabilities.
To evaluate a child with an ocular problem, the ophthalmologist obtains detailed pertinent ocular, developmental, and systemic histories; gestational, natal, and neonatal histories; and any family history of similar or other systemic and ocular diseases. The following examinations should be routinely performed in the pediatric ophthalmologist’s office: (1) estimate of visual acuity in infants and toddlers and exact visual acuity in older children; (2) examination of ocular motility and determination of binocularity of vision; (3) a good anterior segment examination, preferably using the slit lamp; (4) examination of pupillary responses; (5) a dilated fundus inspection using the indirect ophthalmoscope; and (6) a cycloplegic refraction.
DISORDERS APPARENT AT BIRTH OR SHORTLY THEREAFTER
Congenital malformations may be observed at any age. We will only discuss the most common ones and those requiring medical attention or screening for associated systemic abnormalities. Children with any of the following conditions should be referred to the pediatric ophthalmologist.
Birth Trauma
Although birth trauma is not a congenital anomaly per se, it is discussed here because it is evident soon after birth. Forceps injuries to the eye are unilateral and most frequently involve the left eye, probably because the most common position of the infant’s head is left occiput anterior. In these injuries, lid swelling and corneal opacification are apparent soon after birth. Characteristic vertical or oblique breaks in Descemet membrane are observed on slit-lamp examination, in contrast to the horizontal breaks or Haab striae of congenital glaucoma. Myopia, astigmatism, and amblyopia may subsequently develop. More severe injuries can lead to intraocular hemorrhage, breaks in Bruch membrane of the retina, or even rupture of the globe. Prolonged labor and anatomic crowding are predisposing factors.
Adnexal Disorders
In cryptophthalmos, the upper and lower lids are completely fused, although the underlying eyeball may be completely normal. Surgical incision in the area of the palpebral fissure may open directly into the anterior segment of the eye. The cryptophthalmos or Fraser syndrome includes lid fusion, hypertelorism, and cardiac and genital anomalies; it is inherited as an autosomal recessive disorder and results from mutations in the FRAS1 gene, which appears to be important in signal transduction in the extracellular matrix.
 FIGURE 128.1. Midface of patient with Waardenburg syndrome. Note telecanthus and white forelock. |
Upper lid colobomas (i.e., full-thickness defects in lid tissue involving the lid margin) are seen in Goldenhar syndrome, a variant of the oculoauriculovertebral sequence, in conjunction with epibulbar dermoids and preauricular skin tags. Lower lid colobomas are more characteristic of the Treacher Collins syndrome, a form of mandibulofacial dysostosis.
Distichiasis is the growth of true cilia in ectopic locations and in extra rows along the lid margin and out of the orifices of meibomian glands. The distichiasis–lymphedema syndrome is inherited in an autosomal dominant fashion and results from mutations in the FOXC2 gene.
Hypertelorism is a radiologic diagnosis that refers to an increased distance between the two orbits. Telecanthus refers to an unusually long distance between the inner canthi; the ratio between the inter-inner canthal distance and the inter-outer canthal distance is normally about 1:3. Telecanthus and outer displacement of the inferior lacrimal puncti are seen in Type I Waardenburg syndrome (Fig. 128.1), other features of which include heterochromia, deafness, and a white forelock; this syndrome is classified with the neurocristopathies and results from mutations in the PAX3 gene. Hypertelorism occurs in many dysmorphic syndromes, discussed in Chapter 166.
Anterior Segment Disorders
In microcornea, the corneal diameter is 10 mm or less. Microphthalmos may or may not be present.
In microphthalmos, the anteroposterior diameter of the eye is short (normally 18 mm at birth). High hyperopia or myopia is often present. Chorioretinal or iris colobomas and other ocular anomalies may also exist. Microphthalmos may be unilateral, bilateral, isolated, or part of a multisystem dysmorphic syndrome (Box 128.1). Nanophthalmos (i.e., pure microphthalmos) is an autosomal recessive condition involving high hyperopia and a predisposition to narrow-angle glaucoma and spontaneous choroidal effusion.
In megalocornea, the corneal diameter is increased to more than 12.5 mm. The intraocular pressure and endothelial cell count are normal. Megalocornea is usually X-linked recessive and may be rarely seen in Marfan syndrome. Congenital glaucoma should be ruled out in infants with enlarged corneal diameter. Table 128.2 lists the differences between congenital glaucoma and megalocornea.
BOX 128.1 Practical Classification of Microphthalmia/Coloboma
Isolated
Microphthalmia (AD, AR, XR)
Colobomatous
Noncolobomatous (nanophthalmos)
Isolated uveoretinal coloboma
Microphthalmia with cyst
Microphthalmia with ocular anomalies
Microphthalmia with cataract (AD, AR)
Microphthalmia with myopia and corectopia (AD)
Microphthalmia with ectopia lentis
Microphthalmia with congenital retinal detachment (AR)
Persistent hyperplastic primary vitreous (sporadic)
Aicardi syndrome
Microphthalmia with mental retardation
Microphthalmia with mental retardation (AD, AR, XR)
Microphthalmia with mental retardation and congenital spastic diplegia (Sjögren-Larsson syndrome)
Microphthalmia with craniofacial malformations
Facioauriculovertebral sequence
Hallermann-Streiff syndrome
Amniotic band syndrome
Transverse facial cleft
Microphthalmia with cleft lip/palate
Microphthalmia with microcephaly
Microphthalmia with microcephaly and retinal folds (XR, AR)
Microphthalmia with hydrocephalus and congenital retinal nonattachment (Warburg syndrome)
Microphthalmia with malformations of the hands and feet
Microphthalmia with polydactyly
Waardenburg anophthalmia syndrome (AR)
Subgroup of CHARGE association (bifid thumbs)
Microphthalmia with multiple congenital anomalies (syndromes)
CHARGE association
Duker syndrome
Lenz microphthalmia syndrome (XR)
Oculodentoosseous dysplasia (AD, AR)
Cryptophthalmos syndrome (AR)
Cerebrooculofacial syndrome (AR)
Goltz syndrome or focal dermal hypoplasia (XD)
Lowe syndrome (XR)
Meckel-Gruber syndrome (AR)
Basal cell nevus syndrome of Gorlin-Goltz (AD)
Congenital contractural arachnodactyly (AD)
Rubinstein-Taybi syndrome
Cross syndrome (AR)
Fanconi syndrome (AR)
Diamond-Blackfan syndrome (AR)
Epidermal nevus syndrome
Microphthalmia in chromosomal anomalies
T-13 (Patau syndrome)
4p- (Wolf-Hirschhorn syndrome)
18q-
18r
T-18 (Edward syndrome)
Cat-eye syndrome (marker 22)
Other chromosomal aberrations
Microphthalmia and intrauterine insults
Maternal drug intake: thalidomide, alcohol, isotretinoin
Maternal vitamin A deficiency
Maternal fever or radiation exposure
Maternal uncontrolled phenylketonuria
Intrauterine infections: cytomegalovirus, Epstein-Barr virus, varicella, herpes simplex, rubella, toxoplasmosis
Footnote
AD, autosomal dominant; AR, autosomal recessive; XD, X-linked dominant; XR, X-linked recessive.
In sclerocornea, there is loss of transparency of the cornea. This results from irregular arrangement of the collagen fibrils, which are normally arranged in a hexagonal fashion in the transparent cornea. Other ocular abnormalities such as aniridia, cataracts, and coloboma may coexist with sclerocornea. Patients may also have systemic birth defects.
TABLE 128.2. DIFFERENCES BETWEEN MEGALOCORNEA AND CONGENITAL GLAUCOMA | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Anterior segment dysgenesis is a term that encompasses a variety of developmental defects of the anterior chamber angle and iris. These malformations are most commonly caused by abnormal neural crest differentiation into corneal endothelial cells, anterior chamber angle, and anterior iris. In the mildest form called posterior embryotoxon, there is a prominent anteriorly displaced Schwalbe line (i.e., peripheral end of Descemet membrane). In the Axenfeld anomaly, iris strands are attached to the anteriorly displaced Schwalbe line. The Rieger anomaly involves hypoplasia of the anterior stroma of the iris in addition to the abnormalities already described. Anterior segment dysgenesis is inherited in an autosomal dominant fashion. At least three genetic loci have been assigned to syndromes featuring a Rieger ocular phenotype. Developmental of adult open-angle glaucoma occurs in 50% of these patients over the course of their lifetime. The Rieger syndrome combines anterior segment dysgenesis with hypodontia and redundant umbilical skin. It is inherited in an autosomal dominant fashion and its gene, PITX2, has been mapped to 4q23. PITX2 is a homeobox-containing gene and is important in neural crest development.
Peters anomaly involves a central defect in Descemet membrane, with various degrees of adhesion of the iris and lens
capsule to the central cornea; these presumably develop from failure of separation of the lens from the surface ectoderm as the cornea is forming. Cataracts may be present. Combined cataract extraction and penetrating keratoplasty are required in severe cases. Most instances are sporadic, although autosomal recessive and autosomal dominant forms have been observed. Glaucoma is common and is a leading cause of visual loss in these patients. One-quarter to one-fourth of patients with Peters anomaly have other ocular malformations and a similar proportion of patients have other birth defects of the brain, heart, kidneys, and extremities.
capsule to the central cornea; these presumably develop from failure of separation of the lens from the surface ectoderm as the cornea is forming. Cataracts may be present. Combined cataract extraction and penetrating keratoplasty are required in severe cases. Most instances are sporadic, although autosomal recessive and autosomal dominant forms have been observed. Glaucoma is common and is a leading cause of visual loss in these patients. One-quarter to one-fourth of patients with Peters anomaly have other ocular malformations and a similar proportion of patients have other birth defects of the brain, heart, kidneys, and extremities.
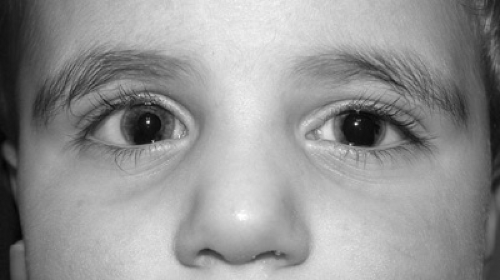 FIGURE 128.2. Patient with aniridia. Note the presence of iris remnants in both eyes, right more than left. |
Uveal Tract Disorders
In aniridia (Fig. 128.2), a rim of rudimentary iris is always present at the iris root. The associated ocular features include cataracts, ectopia lentis, developmental glaucoma, corneal pannus, persistence of the retina over pars plana, and foveal hypoplasia leading to decreased visual acuity and nystagmus. Aniridia can be sporadic or hereditary with autosomal dominant transmission. Sporadically affected family members may have atypical iris defects and pseudopolycoria. Wilms tumor has been associated only with sporadic aniridia, and the Wilms tumor, aniridia, genitourinary abnormalities, and mental retardation (WAGR) syndrome has been associated with a deletion of the short arm of chromosome 11. Aniridia results from mutations of the PAX6 gene located on 11p13.
In ocular melanocytosis or melanosis oculi, there is hyperpigmentation of the uveal tract from increased numbers of normal melanocytes. Uveal malignant melanoma develops in less than 1% of patients. In oculodermal melanocytosis or nevus of Ota, ocular melanocytosis is associated with congenital hyperpigmentation of the skin in the distribution of the trigeminal nerve. Oculodermal melanocytosis is more common in African Americans and in Asians than in Caucasians, but malignant melanoma is less frequent in the former than in the latter.
Persistent strands of the pupillary membrane are commonly seen and result from failure of regression of the embryonic tunica vasculosa lentis that obliterates the pupil in utero. The strands seldom interfere with vision.
Chorioretinal colobomas are defects in the retina and uveal tract caused by failure of the embryonic ocular fissure to close. Typical colobomas are located inferiorly because the embryonic fissure is located inferonasally. The iris, ciliary body, inferior choroid, or optic nerve head may be involved singly or in combination (Fig. 128.3). Eyes with colobomas may be of normal size but are generally microphthalmic. Large colobomas may produce a white reflex from the pupil and have been confused with eyes that harbor retinoblastoma. Colobomas may be isolated, sporadic, or hereditary (most commonly autosomal irregular dominant with reduced penetrance), or they may be part of a complex malformation syndrome of known or unknown cause. Box 128.1 presents an etiologic classification of ocular colobomas and of microphthalmia.
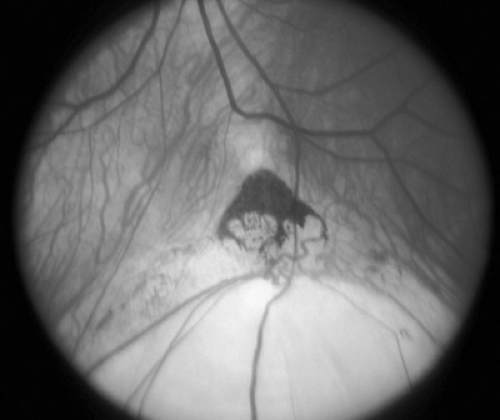 FIGURE 128.3. Typical inferior chorioretinal coloboma along the line of closure of the embryonic fissure. The optic nerve is at the upper edge of the figure. |
Vitreous Disorders
Persistence of hyaloid vessels and vascular loops at the optic disc results from incomplete regression of the hyaloid system of blood vessels. Vision is normal. Rarely, retinal vascular occlusive disease has resulted from twisted prepapillary loops.
Mittendorf dot and Bergmeister papilla are glial remnants of the regressed hyaloid system at the posterior lens capsule and optic disc, respectively. No patent blood vessels are found within these fibrous remnants.
Persistent hyperplastic primary vitreous (PHPV) or persistent fetal vasculature (PFV) is the most severe developmental anomaly that involves the vitreous and the globe. The affected eye is microphthalmic, the ciliary processes are elongated, and there is some degree of anterior or posterior hyperplasia of the fibrous and glial tissue surrounding the hyaloid blood vessels. There may be an associated cataract. PHPV is a unilateral condition affecting male and female patients equally. Visual prognosis depends on the severity of the microphthalmia and the associated retinal traction and dysplasia. The younger the patient is, the more likely it is that surgical intervention to remove the cataract and clear the visual pathways can improve visual potential. After a lensectomy and vitrectomy procedure, immediate aphakic correction and aggressive amblyopia therapy are necessary to produce useful vision. Surgery should be performed before 2 months of age, and parents should be informed about the guarded prognosis. Surgery prevents the later occurrence of angle-closure glaucoma, which otherwise develops in many patients with PHPV. Because it causes a white pupil, PHPV should be differentiated from retinoblastoma on the basis of clinical clues and ultrasonography or computed tomography.
Retinal Disorders
Retinal dysplasia is a pathologic term describing abnormal, disorderly acinar, tubular, and rosette-like formations in the
retina. It is a prominent feature in trisomy 13 and can be produced by intrauterine insults or infection. Retinal dysplasia can lead to the appearance of a white pupil and should therefore be differentiated from retinoblastoma. Retinal dysplasia is also present in patients with Norrie disease, an X-linked condition characterized by congenital blindness, deafness in about 25% of patients, and a progressive neuropsychiatric illness. The gene has been cloned and can be tested for mutations in individuals suspected to have this condition.
retina. It is a prominent feature in trisomy 13 and can be produced by intrauterine insults or infection. Retinal dysplasia can lead to the appearance of a white pupil and should therefore be differentiated from retinoblastoma. Retinal dysplasia is also present in patients with Norrie disease, an X-linked condition characterized by congenital blindness, deafness in about 25% of patients, and a progressive neuropsychiatric illness. The gene has been cloned and can be tested for mutations in individuals suspected to have this condition.
Myelinated nerve fibers are found in about 1% of autopsy eyes. These are present at birth, have a feathery appearance, and are usually found in the nerve fiber layer in contiguity with the optic nerve head, although they can be separate from it. Involvement can be minimal to extensive. In extensive cases a scotoma is produced. There exists a condition in which extensive myelinated nerve fibers are associated with high myopia and amblyopia.
There are several congenital vascular disorders of the retina. Retinal arteriovenous communications are large, dilated retinal vessels. Cavernous hemangiomas, composed of dilated saccular aneurysmal grape-like compartments, are associated with similar central nervous system and cutaneous hemangiomas. These vascular tumors may be inherited in a dominant fashion, and result from mutations in the KRIT1 gene. Coats disease (i.e., congenital retinal telangiectasia) is discussed later in this chapter, in the section on retinoblastoma. Von Hippel-Lindau disease is an autosomal dominant condition characterized by retinal capillary hemangiomas (Fig. 128.4); cerebellar, medullary, and spinal cord hemangioblastomas; and a variety of other cystic and neoplastic lesions throughout the body. The lesions include cysts of the pancreas, kidney, lungs, and ovaries; adenomas of the liver, epididymis, and adrenals; hypernephromas; pheochromocytomas; and familial islet cell tumors. Retinal lesions may be complicated by exudative retinopathy and retinal detachment and have been treated with various degrees of success by laser photocoagulation and cryotherapy. The disease is caused by mutations in the tumor-suppressor VHL gene.
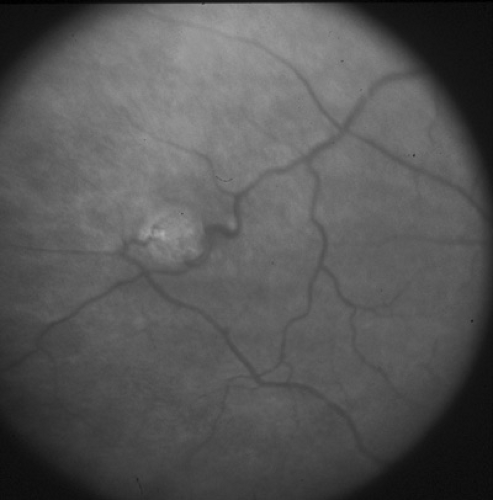 FIGURE 128.4. Small retinal capillary hemangiomas in a patient with von Hippel-Lindau disease. |
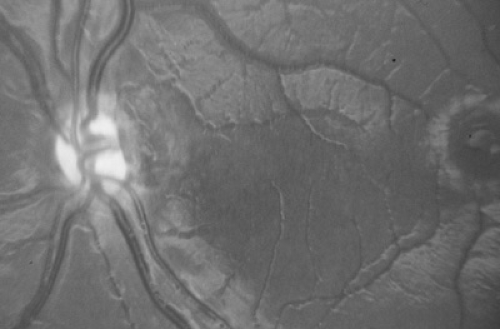 FIGURE 128.5. Very small optic nerve head characteristic of optic nerve hypoplasia. Note increased distance between nerve head on the left and fovea on the right. In normal conditions, one can fit about two optic discs in the area between the disc and the fovea. |
Optic Nerve Disorders
Optic nerve hypoplasia is probably the most common ocular developmental anomaly. It is nonprogressive and characterized by a subnormal number of axons in the affected nerve with normal mesodermal and glial supporting tissues. Subtle, segmental, and severe forms are seen. Classically, the nerve head is one-half to one-third of the normal size, pale to gray, and surrounded by a yellowish halo bordered on either side by a darker ring of pigment, the so-called double ring sign (Fig. 128.5). The retinal vessels are frequently tortuous. Bilateral cases are more frequent than unilateral cases, with variable asymmetry. Visual acuity ranges from poor light perception to normal, and visual field defects are common. Severe bilateral cases are manifested in infancy with poor visual development and nystagmus; less severe and more subtle forms are detected only later in life. Optic nerve hypoplasia has been reported with porencephaly, isolated cerebral atrophy, basal encephaloceles, congenital suprasellar tumors, colpocephaly, and anencephaly. Neuroradiologic abnormalities and hypopituitarism with growth restriction and hypothyroidism appear to be most common in patients with bilateral, severe involvement. Although most cases are sporadic, dominant inheritance has been reported. Maternal diabetes has been associated with an increased incidence of mild optic nerve hypoplasia and good visual acuity. Other rare associations include maternal viral infections and maternal ingestion of quinine and anticonvulsants. Optic nerve hypoplasia may be a common feature of the fetal alcohol syndrome. Other ocular features of the fetal alcohol syndrome are strabismus and a typical configuration of the lids with mild bilateral ptosis and down-slanted and horizontally shortened fissures.
In septooptic dysplasia (De Morsier syndrome), bilateral optic nerve hypoplasia is associated with lack of a septum pellucidum, partial or complete agenesis of the corpus callosum, and dysplasia of the anterior third ventricle. Hypopituitarism may be present. All children with optic nerve hypoplasia should undergo magnetic resonance imaging (MRI) of the brain and pituitary and should have pediatric neurologic and endocrinologic evaluations.
The morning glory disc anomaly (MGDA) is a congenital malformation in which the optic nerve head is excavated, with white tissue at its center and a raised annulus of pigmentary chorioretinal change at its edge (Fig. 128.6). Its appearance and the degree of dysplasia vary. Most cases are unilateral, and visual impairment varies greatly, with vision ranging
from 20/30 to light perception only. Several ocular anomalies may coexist with the disc anomaly. Associated systemic abnormalities include basal encephaloceles, Moyamoya disease, and rare kidney malformations. Infants and children with MGDA should undergo magnetic resonance angiography (MRA) of the carotid circulation to specifically look for vascular abnormalities and Moyamoya vessels. Nonrhegmatogenous retinal detachment occurs in about one-third of cases and is thought to be due to the accumulation of fluid from between the subarachnoid space into the subretinal space through the malformed optic papilla.
from 20/30 to light perception only. Several ocular anomalies may coexist with the disc anomaly. Associated systemic abnormalities include basal encephaloceles, Moyamoya disease, and rare kidney malformations. Infants and children with MGDA should undergo magnetic resonance angiography (MRA) of the carotid circulation to specifically look for vascular abnormalities and Moyamoya vessels. Nonrhegmatogenous retinal detachment occurs in about one-third of cases and is thought to be due to the accumulation of fluid from between the subarachnoid space into the subretinal space through the malformed optic papilla.
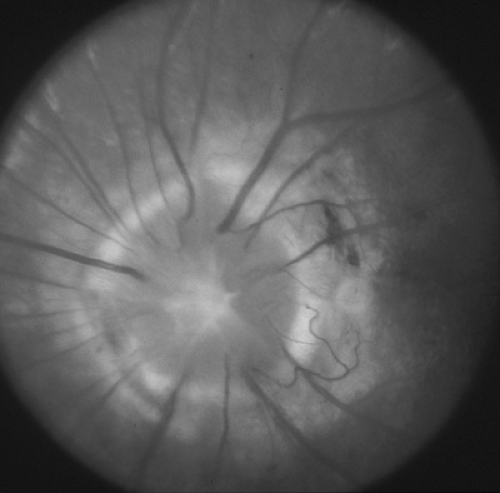 FIGURE 128.6. Morning glory disc anomaly. There is a large opening of the posterior sclera with radial vessels, peripapillary ring of tissue, and central tuft of glial tissue. |
Corneal Opacities
The five categories of disease leading to corneal opacification at birth or in the first few months of life are congenital anomalies, intrauterine and perinatal infections, birth trauma, glaucoma, and corneal dystrophies. Metabolic conditions such as the mucopolysaccharidoses and the mucolipidoses do not give rise to corneal opacification before a few months of age. All children with corneal opacification should be referred to an ophthalmologist.
Leukocoria
Leukocoria (white pupil) is a white or tan reflex in the normally black pupillary area (Fig. 128.7). The reflex, which may be observed in certain ambient lighting conditions or only in certain directions of gaze, could theoretically result from opacification or tumefaction of any structure behind the iris (e.g., lens, vitreous, retina, choroid). Box 128.2 lists conditions that may be associated with leukocoria. The exact cause of the white reflex should be determined as soon as possible so that treatment for the underlying disease can be started. Retinoblastoma, discussed elsewhere in this chapter and in Chapter 101, is a major concern for children with leukocoria.
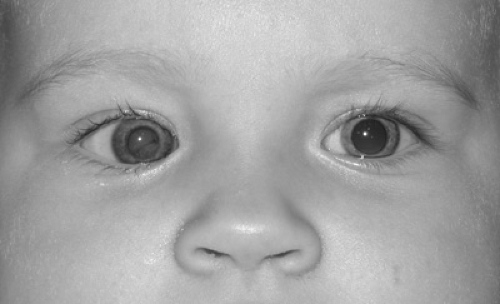 FIGURE 128.7. Dimmer, light orange or tan reflex from right eye as opposed to red reflex from left eye. See Color Figure 128.7 in color section. |
Retinopathy of Prematurity
Retinopathy of prematurity (ROP) is a vasoproliferative retinal disease that occurs in premature infants exposed to high concentrations of oxygen for prolonged periods. Two phases are observed: an acute proliferative phase and a cicatricial phase in which scarring and traction retinal detachment occur. More than 90% of patients with acute disease undergo spontaneous regression, and fewer than 10% of eyes develop significant cicatrization. It is estimated that about 40,000 premature infants are born annually in the United States, and that about 5% of these babies develop some degree of cicatricial ocular damage. Of the latter group, only 5% are totally blind or have severe permanent visual impairment. The most important risk factor in the development of ROP is low birth weight. The disease is rare in infants who weigh more than 2000 g at birth. ROP develops in 2% to 20% of babies weighing between 1,000 g and 1,500 g, and in 30% to 40% of those weighing less than 1,000 g. Other risk factors include gestational age, duration and concentration of oxygen exposure, shift of the oxygen dissociation curve by transfused adult hemoglobin, sepsis, high light intensity, hypoxia, and hypothermia.
Retinal vessels start growing at the nerve head from hyaloid vessels at 4 months’ gestation and progress centrifugally to reach the nasal retina by 8 months and the temporal retinal periphery by 9 months or shortly after birth. ROP results from incomplete vascularization and sprouting of new vessels from the demarcation line between vascularized and nonvascularized retina. The pathogenic mechanisms of new vessel formation and the roles of the various agents implicated in ROP have not been fully elucidated; hypoxemia and hyperoxic damage to growing retinal vessels seem to be important factors. Fibrovascular proliferation results in traction on the normal retina, dragging of the macula and disc, and in partial or total retinal detachment in severe cases. In progressive ROP, the iris is involved, and dilated iris vessels can be seen on anterior segment examination.
An international committee developed a staging classification of ROP. In stage I, there is a demarcation line between vascular and avascular retina. In stage II, there is thickening of the demarcation line and formation of an intraretinal ridge. In stage III, new vessels arise from the ridge, and hemorrhages may be seen on or adjacent to the ridge. In stage IV, subtotal retinal detachment occurs posterior to the ridge, possibly involving one or more quadrants with traction and/or exudative components; the detachment is extrafoveal in stage IV-A and involves the fovea in stage IV-B. In stage V, there is total funnel-shaped retinal detachment, the funnel taking on one of various
configurations. Dilated posterior pole vessels place the disease level at the “plus” level, and the disease becomes more likely to progress to more advanced stages. The various stages can be localized to one of three zones of posteroanterior involvement. Neovascularization in stage III is quantified by clock hours of circumferential involvement. The more posterior the separation between vascularized and nonvascularized retina is, the lower the zone is. The more clock hours of neovascularization there are and the lower (or more posterior) the zone of the disease process is, the more likely is the progression to a poor visual outcome.
configurations. Dilated posterior pole vessels place the disease level at the “plus” level, and the disease becomes more likely to progress to more advanced stages. The various stages can be localized to one of three zones of posteroanterior involvement. Neovascularization in stage III is quantified by clock hours of circumferential involvement. The more posterior the separation between vascularized and nonvascularized retina is, the lower the zone is. The more clock hours of neovascularization there are and the lower (or more posterior) the zone of the disease process is, the more likely is the progression to a poor visual outcome.
BOX 128.2 Differential Diagnosis of a White Pupillary Reflex (Leukocoria)
Hereditary conditions
Norrie disease
Congenital cataract
Coloboma
Congenital retinoschisis
Incontinentia pigmenti
Familial exudative vitreoretinopathy
Developmental anomalies
Posterior hyperplastic primary vitreous
Cataract
Coloboma
Retinal dysplasia
Congenital retinal fold
Myelinated nerve fibers
Morning glory disc anomaly
Congenital corneal opacities
Inflammatory conditions
Nematode endophthalmitis (toxocariasis)
Congenital toxoplasmosis
Congenital cytomegalovirus retinitis
Herpes simplex retinitis
Peripheral uveoretinitis
Metastatic endophthalmitis
Orbital cellulitis
Tumors
Retinoblastoma
Retinal astrocytoma
Medulloepithelioma
Glioneuroma
Choroidal hemangioma
Retinal capillary hemangioma
Combined retinal hamartoma
Miscellaneous conditions
Retinal telangiectasia with exudation (Coats disease)
Retinopathy of prematurity
Rhegmatogenous retinal detachment
Vitreous hemorrhage
Perforating ocular injuries
Battered child syndrome
Footnote
Modified from
Shields JA, Augsburger JJ. Current approaches to the diagnosis and management of retinoblastoma. Surv Ophthalmol 1981;25:347.
Newborns at risk for ROP should be examined by an ophthalmologist after discontinuation of oxygen therapy and before hospital discharge. If ROP is discovered, examinations should be repeated frequently. Significant changes may occur within days in patients with stage III disease, and laser therapy may be indicated. Most retina specialists have abandoned cryotherapy and favor the use of laser ablation in ROP. If regression is documented, examinations are done less frequently. The optimal time for examination is 6 to 10 weeks postpartum because this is when most cases of ROP are detected. Long-term complications of regressed ROP include high myopia and angle-closure glaucoma.
Current treatment of ROP consists of laser surgery to the avascular retina to arrest progression of the disease. A large multicenter trial has documented the value of this therapy. ROP has to reach, in general, a threshold of “plus” disease with 5 contiguous clock hours of neovascularization or 8 noncontiguous clock hours of neovascularization before treatment is initiated. However, more recent studies suggest that earlier treatment may be beneficial to selected patients, especially those with zone 1 and plus disease. Laser treatment appears to be better tolerated by infants and achieves similar favorable outcome as compared to the more invasive and less–well-tolerated cryotherapy. Vitrectomy and scleral buckling are performed in stages IV and V of the disease; visual results are variable but some patients achieve ambulatory or better vision. Intravenous vitamin E supplementation is not helpful in the prevention of ROP in premature infants and may be associated with a higher than normal risk of intraventricular hemorrhage and other complications of prematurity.
COMMON EYE PROBLEMS
Errors of Refraction
The most common cause of poor vision in childhood and adolescence is an error of refraction, the presence of which can be determined accurately by retinoscopy. The indications for prescribing glasses in children include significant visual impairment that interferes with the child’s activity or the presence of asthenopia (ocular strain), strabismus, anisometropia (unequal error of refraction between the two eyes), or high astigmatism. The latter three conditions all predispose to amblyopia.
Visual Impairment
The pediatrician or ophthalmologist may be faced with the very delicate situation of parents observing poor visual responsiveness in their baby; the situation is even more complex in the case of a concomitant developmental delay or associated systemic disease. Before any statement is made about the infant’s visual status or prognosis, a pediatric ophthalmologist should be consulted to perform a thorough evaluation. The clinical assessment includes observing the infant’s general responsiveness to visual stimuli, recording abnormal ocular movements, and documenting wandering conjugate eye movements, which in blind children are usually horizontal and roving with or without tonic spasms and vertical jerky movements. Strabismus may or may not be present, and pupillary responses to a bright light stimulus may be reduced from normal levels. A careful search
for organic eye disease and developmental anomalies is made, and refraction is tested. Optokinetic testing and cortical visual-evoked responses are helpful in documenting the presence of vision. Electroretinography should be performed for children in whom blindness is strongly suspected but who have a normal ocular examination and no evidence of cortical visual impairment. This test allows the detection of Leber congenital amaurosis, an autosomal recessive retinal dystrophy with onset at birth and in which visible retinal abnormalities may be absent or minimal.
for organic eye disease and developmental anomalies is made, and refraction is tested. Optokinetic testing and cortical visual-evoked responses are helpful in documenting the presence of vision. Electroretinography should be performed for children in whom blindness is strongly suspected but who have a normal ocular examination and no evidence of cortical visual impairment. This test allows the detection of Leber congenital amaurosis, an autosomal recessive retinal dystrophy with onset at birth and in which visible retinal abnormalities may be absent or minimal.
Unilateral visual loss is more difficult to detect. Affected infants or children usually present with strabismus or with leukocoria, or the condition may be discovered on routine examination. Causes of unilateral blindness include unilateral high errors of refraction; various congenital abnormalities of the eye, especially optic nerve hypoplasia; trauma; and rarely retinoblastoma. All efforts should be made to uncover the cause of severe visual impairment in infants and children so that appropriate therapy can be instituted early and so that prognosis and genetic counseling can be offered in cases of inherited diseases with ocular involvement.
Leber congenital amaurosis is characterized by moderately to severely reduced vision before the age of 1 year, poor pupillary reaction, retinal degeneration, and markedly reduced or extinguished electroretinographic waveform. The disease is autosomal recessive, but there is genetic heterogeneity and several genetic defects have been identified to date. The ophthalmoscopic appearance is variable, ranging from normal to a typical retinitis pigmentosa-like picture with bone spicule formation, attenuation of retinal vessels, and waxy optic atrophy. Associated retinal changes include a salt-and-pepper appearance, chorioretinal atrophy, macular colobomas, retinitis punctata albescens, disc edema, and a nummular pigmentary pattern with round to oval pigmented lesions. Affected eyes may develop cataracts or keratoconus when the affected person is in his or her teens or 20s. Congenital cataracts are rare. Infants with Leber congenital amaurosis are usually examined in the first year of life because of poor vision, wandering eye movements, photophobia, and the oculodigital sign (i.e., the infant rubs the blind eyes, probably in an attempt to elicit some visual images through mechanical excitation of the retina). Although ophthalmoscopic findings change with age, visual acuity remains stable except in a subgroup of patients with macular “colobomas,” in whom vision deteriorates. Associated systemic abnormalities include polycystic kidney disease, osteopetrosis, and skeletal anomalies. Neuropsychiatric disorders and mental retardation may coexist in patients with Leber congenital amaurosis and may be associated with concomitant central nervous system malformations or disease. The electroretinogram is essential to the diagnosis of this condition. Other causes of poor vision in infancy include bilateral optic nerve hypoplasia, achromatopsia, albinism, aniridia, congenital stationary night blindness, macular coloboma, and infectious chorioretinitis. Systemic diseases with retinal findings similar to those of Leber congenital amaurosis include Senior syndrome (familial nephronophthisis and tapetoretinal degeneration); Saldino-Mainzer syndrome (Senior syndrome plus cone-shaped epiphyses of the hands); Bassen-Kornzweig syndrome (abetalipoproteinemia); several peroxisomal disorders such as neonatal adrenoleukodystrophy, Zellweger syndrome, and infantile phytanic storage disease; and neurodegenerative disorders such as infantile neuronal ceroid lipofuscinosis. Peroxisomal disorders should be suspected in infants with pigmentary retinopathy, cataracts, hypotonia, and a Zellweger phenotype. Recent therapeutic interventions in animal RPE65 models of Leber congenital amaurosis promise effective intervention in some patients with this devastating disease.
Retinitis pigmentosa is a group of hereditary diseases characterized by progressive degeneration of the retina, retinal pigment epithelium, and choroid, with resultant loss of visual field and acuity. Ophthalmoscopy characteristically shows thinning of retinal vessels, waxy pallor of the optic disc, and peripheral bone corpuscle pigmentary changes, first in the equatorial area (Fig. 128.8). Choroidal sclerosis is a late feature of the disease. As the condition progresses, a peripheral scotoma appears, enlarges, and eventually reduces the visual field to a central area where acuity may be well preserved. Night blindness is a universal finding. Diagnosis is made clinically and confirmed on electroretinography. The electroretinographic response is markedly subnormal or nonrecordable, even in the absence of subjective visual symptoms. Other ocular findings include posterior subcapsular cataracts, myopia, keratoconus, and vitreous degeneration with a cellular response. About 25% of families with autosomal dominant retinitis pigmentosa have mutations in the rhodopsin gene. Others have mutations in the peripherin gene or other molecules involved in the process of phototransduction. Three modes of inheritance are recognized. X-linked recessive and autosomal recessive retinitis pigmentosa are more severe, start earlier, and result in earlier blindness than the autosomal dominant form. Nongenetic cases exist, confounding determination of the cause.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


