Ovarian Sex Cord–Stromal Tumors
DAVID M. GERSHENSON  SEAN C. DOWDY
SEAN C. DOWDY  ROBERT H. YOUNG
ROBERT H. YOUNG
The intraovarian matrix that supports the germ cells and is covered by the surface epithelium consists of cells originating from the sex cords and mesenchyme of the embryonic gonad. Granulosa cells and Sertoli cells, generally considered to be homologous, are derived from the sex cord cells, whereas the pluripotential mesenchymal cells are the precursors of the theca cells, Leydig cells, and fibroblasts. Neoplastic transformation of these cellular constituents, either singly or in various combinations collectively, results in neoplasms that are termed sex cord-stromal tumors (SCSTs). The classification of the SCSTs provides the template from which this chapter endeavors to stratify and define these tumor entities according to their morphologic characteristics (Table 26.1).
The SCSTs are estimated to account for approximately 7% of all malignant ovarian neoplasms (1). Although SCSTs account for a decreasing proportion of all ovarian malignancies with advancing age, the annual age-related incidence continues to increase through the seventh decade of life (2). Overall, the majority of these tumors are benign or of low malignant potential and are associated with a favorable long-term prognosis. In addition, a significant proportion of SCSTs are diagnosed in patients younger than age 40 years and have the potential to produce a variety of steroid hormones. Hence, adequate knowledge of the natural history of each of these tumors is imperative to diagnose and individualize appropriately definitive surgical and adjuvant therapy.
SCSTs account for nearly 90% of all functioning ovarian neoplasms (3). With the exception of fibromas, the clinical presentation of patients with SCSTs is frequently governed by the clinical manifestations resulting from the endocrinologic abnormalities. Excessive estrogen production, whether from increased tumor synthesis or peripheral conversion of androgens, influences end-organ responses, which are usually age-dependent and can range from isosexual precocious puberty to menometrorrhagia to postmenopausal bleeding. In addition, the associated risks for endometrial cancer and possibly breast cancer must be recognized (4–6). Conversely, the rapid onset of signs ranging from early defeminization to frank virilization heralds a hyperandrogenic state. Elevated circulating levels of testosterone and/or androstenedione provide strong evidence for the presence of an SCST. Although granulosa cell, theca cell, and Sertoli cell tumors are generally considered to be estrogenic, and Sertoli-Leydig cell and steroid cell tumors are predominantly androgenic, the functional endocrinologic capacities of these tumors are impossible to predict based on their morphologic features. It should also be noted that miscellaneous ovarian tumors, both primary and metastatic, that are not in the SCST family may be androgenic or estrogenic if their stroma is stimulated to undergo luteinization.
GRANULOSA CELL TUMORS
Although granulosa cell tumors (GCTs) of the ovary were initially described by Rokitansky in 1859 (7), the etiopathogenesis of these neoplasms remains ill defined. At least in part, this is a reflection of the low incidence of GCTs, and hence the limited number of cases managed at any single institution. Although molecular aberrations crucial to the pathogenesis of GCTs have been elucidated recently, to our knowledge there are no recognized risk factors for their development (4,8). Reproductive factors, including the use of fertility-promoting agents and oral contraceptives, do not correlate consistently with the development of disease. Unkila-Kallio et al. (9) studied a possible link between fertility-promoting agents and GCTs using the nationwide Finnish Cancer Registry. They analyzed the occurrence of GCTs in Finland during the time period 1965 to 1994 against sales statistics for ovulation inducers. The incidence of GCTs declined by nearly 40% from 1965–1969 to 1985–1994 despite a 13-fold increase in the use of clomiphene citrate and a 200-fold increase in human menopausal gonadotropin use; oral contraceptive use increased fivefold. No hereditary predisposition for any of the SCSTs has been identified (10). Of note, a recent case report described the occurrence of adult GCTs in 2 first-degree relatives (11).
GCTs comprise 5% of all ovarian malignancies, but account for approximately 70% of malignant SCSTs (4–6, 12–19). The annual incidence of GCTs in the United States and other developed countries varies from 0.4 to 1.7 cases per 100,000 women (5,6,8,18,20,21). Quirk and Natarajan reported histology-specific age-adjusted ovarian cancer incidence rates that were standardized to the recently adopted year 2000 U.S. standard population (22). They utilized data gathered from the Surveillance, Epidemiology, and End Results (SEER) Program for the years 1992–1999. Out of a total of 23,484 microscopically confirmed cases of primary ovarian cancer, 293 (1.2%) were of sex cord–stromal origin. Although GCTs have been diagnosed from infancy through the tenth decade of life, the peak incidence for these tumors occurs during the perimenopausal decade. The average age at the time of diagnosis in over 750 cases was 52 years (4–6,13,15–18). Considering that GCTs occurring after the third decade of life appear to be histologically distinct in most instances from those occurring in children and younger adults, the clinical and pathologic characteristics for the juvenile and adult GCTs will be addressed separately.
Classification of Sex Cord–Stromal Tumors |
Granulosa-Stromal Cell Tumors
Granulosa cell tumor
Adult type
Juvenile type
Tumors in the thecoma-fibroma group
Thecoma
Fibroma-fibrosarcoma
Sclerosing stromal tumor
Sertoli-Stromal Cell Tumors
Sertoli cell tumor
Leydig cell tumor
Sertoli–Leydig cell tumor
Well differentiated
Of intermediate differentiation
Poorly differentiated
With heterologous elements
Retiform
Mixed
Sex Cord Tumor with Annular Tubules
Unclassified
Gynandroblastoma
Steroid cell tumors
Stromal luteoma
Leydig cell tumor
Hilus cell tumor
Leydig cell tumor, nonhilar type
Steroid cell tumor not otherwise specified
Granulosa Cell Tumors: Adult Type
Adult-type granulosa cell tumors (AGCTs), as histologically described below, account for 95% of all GCTs. The majority of patients will present with one or a combination of the following clinical symptoms: abnormal vaginal bleeding, abdominal distention, and abdominal pain (5,6,15–18,23). The latter symptoms are most frequently attributable to the gross size of the tumor at the time of diagnosis, with the majority exceeding 10 cm in diameter and many exceeding 15 cm (5,16,18). In one series, 12% had ascites at diagnosis (23). In many series, menometrorrhagia, oligomenorrhea, or amenorrhea in pre-menopausal women or bleeding in postmenopausal women is the most common reason for seeking medical assistance. These and other clinical manifestations such as breast tenderness, uterine myohypertrophy, and endometrial hyperplasia are consistent with the presence of an estrogen-secreting tumor.
The endocrine function of AGCTs, specifically the production of estrogens, has been repeatedly demonstrated by assessment of the end organ, the endometrium, and measurements of peripheral levels of estrogen before and after surgery. In a detailed retrospective analysis of endometrial specimens from patients (n = 69) with GCTs, Gusberg and Kardon (24) observed histologic features consistent with unopposed estrogen, including atypical adenomatous hyperplasia in 42% of the evaluated cohort, adenocarcinoma in situ (4) in 5%, and invasive adenocarcinoma in 22%. Similarly, Evans et al. (4) noted endometrial hyperplasia in 55% and adenocarcinoma in 13% of their GCT study population. Other investigators have corroborated the high prevalence of glandular hyperplasia and have reported adenocarcinoma frequencies ranging from 3% to 27% (5,6,13,15–18,25,26). Selective ovarian venous catheterizations during surgery have documented hormonal production, including the secretion of large quantities of estrogen from the ovary harboring the GCT. The return of serum estrogen to physiologic levels after definitive treatment has been witnessed repeatedly. Occasionally, patients with GCTs present with endometrial changes (decidual reaction of the stroma or secretory characteristics of the glands) consistent with tumor production of progesterone (27). Rarely, virilizing changes such as oligomenorrhea, hirsutism, and other masculinizing signs may accompany GCTs (28–30).
Pathology
AGCTs have an average diameter of approximately 12 cm, but a subset, 10%–15% of the cases, are small and not appreciated on pelvic examination (31). Most characteristically, they are predominantly cystic, with numerous locules filled with fluid or clotted blood and separated by solid tissue (Fig. 26.1), or they are solid, with large areas of hemorrhage. The solid tissue may be gray-white or yellow and soft or firm. A rare tumor is cystic, usually thin walled, but occasionally thick walled, and multilocular or unilocular (29).
Microscopic examination reveals an almost exclusive population of granulosa cells or, more often, an additional component of theca cells, fibroblasts, or both. The granulosa cells grow in a wide variety of patterns. The better-differentiated tumors usually have microfollicular, macrofollicular, insular, or trabecular patterns. The microfollicular pattern is characterized by numerous small cavities (Call-Exner bodies) (Fig. 26.2) that may contain eosinophilic fluid, one or a few degenerating nuclei, hyalinized basement-membrane material, or, rarely, basophilic fluid. The microfollicles are typically separated by well-differentiated granulosa cells that contain scanty cytoplasm and pale, angular or oval, often grooved nuclei arranged haphazardly in relation to one another and to the follicles. The uncommon macrofollicular pattern is characterized by cysts lined by well-differentiated granulosa cells beneath which theca cells are present. The trabecular and insular forms of GCTs are characterized by bands and islands of granulosa cells separated by fibromatous or the-comatous stroma. The less well-differentiated forms of the adult granulosa cell tumor typically have a water silk (moire silk), gyriform, or diffuse (sarcomatoid) pattern alone or in combination. The first 2 patterns are manifested by parallel undulating or zigzag rows of granulosa cells, generally in single file, whereas the diffuse form is characterized by a monotonous, patternless cellular growth. In some adult granulosa cell tumors, the neoplastic cells have moderate to abundant quantities of dense or vacuolated cytoplasms; the term luteinized granulosa cell tumor is appropriate when such cells predominate (27). The cells in GCTs usually have round to oval, pale, and often grooved nuclei (Fig. 26.2), but rarely the cells are spindle shaped, resembling a cellular fibroma or low-grade fibrosarcoma; mitotic figures may be numerous, but are rarely atypical. There is usually only mild nuclear atypia, but approximately 2% of tumors contain mono-nucleate and multinucleate cells with large, bizarre, hyperchromatic nuclei, the presence of which does not appear to worsen the prognosis (32).

FIGURE 26.1. Granulosa cell tumor. The sectioned surface is composed predominantly of multiple cysts filled with blood.
Source: Reprinted with permission from Case Records of the Massachusetts General Hospital, Case 89–1961. N Engl J Med. 1961;265:1210–1214.
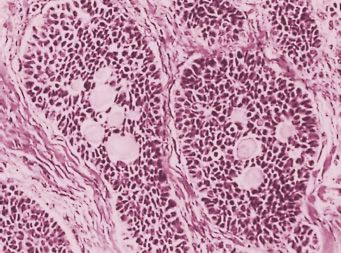
FIGURE 26.2. Granulosa cell tumor, adult type, microfollicular pattern. Several nests of granulosa cells with small oval and angular nuclei enclose multiple Call-Exner bodies.
Natural History
Adult granulosa cell tumors are low-grade malignancies with a propensity to remain localized and demonstrate indolent growth. Ninety percent are stage I at diagnosis (26). The 10-year survival rate for stage I disease ranges from 86% to 96%; for more advanced disease at diagnosis, 26% to 49% (26). Bilaterality occurs in less than 10% (23). Tumor rupture occurred in 22% of a series of 97 cases (23). A unique feature of GCTs is recurrences at extended time intervals from primary therapy, suggesting the presence of persistent occult disease with a very indolent growth rate. For patients who recur, the median time to recurrence is 6 years and median survival after recurrence is 5.6 years (4,13). Numerous reports exist of recurrences occurring more than a decade following primary treatment (33,34).
Prognostic Factors
The staging system for GCTs is the same as that used for epithelial ovarian cancer (International Federation of Gynecology and Obstetrics [FIGO]). Whereas surgical stage has been recognized as the most important prognostic factor for GCTs, the impact of tumor size, rupture, histologic subtype, nuclear atypia, and mitotic activity on outcome is less clear and larger, well-characterized series are necessary to clarify existing discrepancies (8,35,36). As noted above, GCTs are large and therefore prone to rupture. Rupture appears to adversely impact survival in stage I patients, justifying stratification as stage IC (13). However, the prognostic importance of positive cytology and surface involvement is less defined in stage I GCTs (8). In a series of 176 GCTs, only residual tumor after surgery (a surrogate for stage) and tumor size >13.5 cm were associated with recurrence on multivariate analysis (37). In other reports, tumor size lost independent predictability when stratified by stage (5,14,18,20,37). Increasing degrees of nuclear atypia and increasing mitotic frequency per 10 high-power fields (HPFs) have been correlated inversely with prognosis. Specimens from patients with more advanced disease tend to demonstrate higher grade of atypia and/or more mitotic figures (5,13,14,18). Despite its somewhat subjective assessment, nuclear grade has been reported to be a reliable prognostic indicator in stage I cases (13,18). The significance of histologic subtypes and ploidy status has been debated, and they appear to be of minimal value. Several investigative groups (4,5,12–14,18) have failed to confirm Kottmeier’s (38) report of the prognostic importance of histologic patterns alone in GCTs. Similarly, the results of investigations utilizing flow cytometric analysis of DNA content have been inconsistent. Klemi et al. (39) reported a significant survival advantage for patients with tumors demonstrating normal ploidy and/or an S-phase fraction of less than 6%. However, other investigators have suggested that nondiploid GCTs are infrequently encountered (40,41). Chadha et al. (40) reported that 3 of 5 aneuploid tumors from a total population of 43 pathologically diagnosed GCTs were vimentin negative but positive for cytokeratin and epithelial membrane antigen, and therefore cautioned that such highly aneuploid tumors may represent undifferentiated carcinomas. Indeed, it is clear that some series of GCTs in the literature include undifferentiated carcinomas not otherwise specified, or recently recognized entities such as the large-cell carcinoma of hypercalcemic type. Therefore, series with unusually large numbers of late-stage or poor-prognosis cases should be evaluated cautiously.
Investigators have analyzed several potential molecular markers, including p53 status, telomerase, Ki-67, c-myc, HER2/neu, and VEGF in GCTs (42–47). To date, no molecular marker provides prognostic information for GCTs beyond what is known from stage and histopathologic parameters.
Ala-Fossi et al. stained 30 GCTs for the inhibin subunit. All 24 stage I and II tumors were positive, whereas 4 of 6 stage II–IV tumors were negative. Those that were negative were poorly differentiated and exhibited rapid disease progression. Whether other observers would have accepted these tumors as valid GCTs is a concern. Stage was the sole independent prognostic factor (48).
Serum Markers
Recognizing that the majority of patients presenting with advanced GCTs will recur, the identification of a specific serum tumor marker(s) would facilitate early detection of recurrent disease and monitoring of treatment effectiveness (4,5,16,18). As noted above, serum estrogens are generally produced by GCTs and have been utilized as an indicator of disease status (49). Unfortunately, serum estradiol levels are occasionally normal, and more frequently are only marginally increased, making them less than ideal for monitoring in a significant number of patients. Several proteins derived from granulosa cells, including inhibin, follicle-regulating protein, and müllerian-inhibiting substance, are readily assayable in serum and forwarded as useful diagnostic monitoring markers (50–58). In a prospective evaluation of 27 patients with GCTs, Jobling et al. (52) demonstrated that serum inhibin levels are typically elevated sevenfold above normal follicular phase levels prior to primary surgical management. Mom et al. showed that inhibin B was the predominant form of inhibin secreted by these tumors, with a sensitivity and specificity of 89% and 100% compared to 67% and 100% for inhibin A. Elevations in inhibin B were present in 85% of recurrences and predated clinical evidence of recurrence by a median of 11 months (59). In recent years, inhibin and calretinin have become useful immunohistochemical markers to assist in the diagnosis of GCTs and, for that matter, other SCSTs (60).
Granulosa Cell Tumors: Juvenile Type
Ovarian neoplasms are relatively rare in childhood and adolescence. When encountered, the majority are of germ cell origin, with only 5% to 7% being SCSTs. The latter, consisting predominantly of the granulosa cell type in this age group, demonstrates a distinct tumor biology from the typical granulosa cell tumor (AGCT) considered above (61). Approximately 90% of the GCTs diagnosed in prepubertal girls and in most women less than 30 years of age will be of the juvenile type (JGCT). In a clinicopathologic analysis of 125 cases of JGCT, 44% occurred prior to age 10 years and only 3% after the third decade of life (62). The majority of prepubertal patients present with clinical evidence of isosexual precocious pseudopuberty, which may include breast enlargement, development of pubic hair, increased vaginal secretions, advanced somatic development, and other secondary sex characteristics (62–66). Serum estradiol levels were reported as being elevated in 17 of 17 cases of JGCTs with pseudopuberty (64). In addition, elevated levels of serum progesterone (6 of 10) and testosterone (6 of 8) were likewise observed, as well as suppressed levels of luteinizing hormone and follicle-stimulating hormone. The occasional patient will harbor an androgen-secreting JGCT accompanied by virilization (62,64,65). Although the signs of either precocious pseudopuberty or virilization are dramatic, the most consistent clinical sign at presentation in patients with JGCTs is increasing abdominal girth. Young et al. (62) indicated that in only 2 of 113 nonpregnant patients with JGCTs was the treating physician unable to palpate a mass on abdominal, pelvic, and/or rectal examination. Abdominal pain, dysuria, and constipation may coexist. Infrequently, a surgical emergency is encountered following spontaneous rupture or torsion of the enlarged ovary. Juvenile granulosa cell tumors may occur in infants, who appear to have a more favorable prognosis than older individuals (67). Hasiakos et al. described a recent case of JGCT associated with pregnancy and reviewed the literature (68).
The frequency of bilaterality for JGCTs is estimated to be 5%, similar to AGCTs (69). When stage was assigned based on surgical and histologic parameters, 88% were stage IA, 2% stage IB, 8% stage IC, and 3% stage II. As noted, extraovarian spread is infrequently encountered at exploration, whereas rupture of the tumor is noted in approximately 10% of cases. Ascites contributes to the abdominal distention in 10% to 36% of cases (62,64).
JGCTs have been reported in association with enchondromatosis alone (Ollier’s disease) or concomitantly with hemangiomas (Maffucci’s syndrome) (64,70–72). Individuals with these relatively uncommon mesodermal dysplasias generally present prior to puberty and frequently develop secondary neoplasms, most commonly sarcomas, after the second decade of life. Juvenile granulosa cell tumors are the next most frequent tumor associated with these disorders and become evident during the first and second decades of life. These observations appear to imply more than coincidental occurrences and suggest a generalized mesodermal dysplasia, perhaps contributing to the pathogenesis of these neoplastic processes. In addition, congenital bilateral JGCTs of the ovary have been reported in leprechaunism, a disease characterized by insulin resistance resulting from an insulin-receptor defect (73).
Pathology
The appearances of JGCTs are similar to the adult form; a solid and cystic neoplasm, in which the cysts contain hemorrhagic fluid, is common (62,63,66). Uniformly solid and uniformly cystic neoplasms are also encountered; the latter may be multilocular or, in rare instances, unilocular. The solid component is typically yellow-tan or gray and occasionally exhibits extensive necrosis, hemorrhage, or both.
Microscopic examination typically reveals a predominantly solid cellular tumor with focal follicle formation, but occasionally, a uniformly solid or a uniformly follicular pattern is seen. In the solid areas, the neoplastic cells may be arranged diffusely or as multiple nodules of various sizes. The follicles typically vary in size and shape; Call-Exner bodies are rarely encountered, and the follicles rarely reach the large size of those in the macrofollicular AGCT. The follicular lumens in the juvenile tumor contain eosinophilic or basophilic fluid, which stains with mucicarmine in approximately 2 of 3 cases.
The 2 characteristic cytologic features of the neoplastic juvenile granulosa cells that distinguish them from those of AGCT are their generally rounded, hyperchromatic nuclei, which almost always lack grooves, and their almost invariable moderate to abundant eosinophilic or vacuolated (luteinized) cytoplasm (Fig. 26.3). Nuclear atypia in JGCTs varies from minimal to marked; in approximately 13% of the cases, severe degrees are present. The mitotic rate also varies greatly but is generally higher than that seen in AGCTs, often being 5 or more per HPF (62,63).
Natural History
In the initial series by Young et al., 98% of 125 patients with JGCTs were less than 35 years of age, and 78% were 20 years or less (62). Notwithstanding the customary presenting complaint of increased abdominal girth and the clinical documentation of a large mass (64% >10 cm), 90% of the JGCTs analyzed by Young et al. (62) were stage IA or IB. The corresponding survival rate for these patients with an average follow-up of 3.5 years was 97%. Included were 9 stage IA2 patients with rupture of the tumor, all of whom were alive and free of disease. Patients presenting with associated isosexual pseudoprecocious puberty may have a more favorable prognosis. Assessing 80 such cases from 212 reported JGCTs, only 2 cancer-related deaths (2.5%) were observed. Presumably, the clinical manifestations lead to early diagnosis and excellent outcomes (62,64–66).
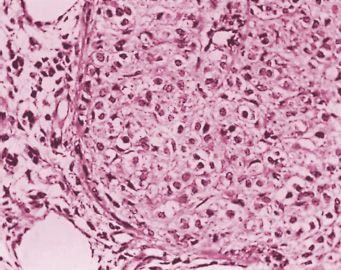
FIGURE 26.3. Granulosa cell tumor, juvenile type. A nodule of tumor is composed of large cells with abundant cytoplasm and slightly pleomorphic, hyperchromatic nuclei.
Although the presentation of early symptoms and localized disease are similar to AGCTs, the natural history of JGCTs differs notably in several respects. Although the adult form frequently includes a latency period with recurrences remote from initial diagnosis, the juvenile counterpart is characteristically aggressive in advanced stages and the time to relapse of limited duration, generally within 3 years of initial diagnosis (62,64,66). Thirteen cases of stage II, III, or IV disease were abstracted from 3 analyses with a combined sample size of 180 patients (62,64,65). Of these 13 cases, only 3 patients (23%) were alive when reported, and notably, the recurrences and deaths occurred within a relatively brief interval.
Prognostic Factors
Young et al. (62) noted surgical stage to represent the most reliable prognostic indicator. Tumor size, mitotic activity, and nuclear atypia were significant predictors only when analyzed without regard to stage. In that series, rupture did not correlate with outcome. Schneider et al. reported on a group of 54 SCSTs in children and adolescents from Germany (45 JGCTs and 9 others) (74). They addressed the outcome of patients with “accidental” stage IC disease, defined as violation of the tumor capsule during surgery, versus “natural” stage IC tumors, with preoperative rupture or malignant ascites. Among 12 patients with accidental stage IC disease, there were no recurrences. In contrast, 5 of the 9 patients with natural stage IC disease recurred (p = 0.001). Assessment of DNA content via flow cytometry in JGCTs demonstrated nondiploid patterns in nearly half (75,76). However, Jacoby et al. (75) were unable to correlate DNA ploidy or S-phase fraction (SPF) with either stage of disease or prognosis in patients with localized disease. In the series by Schneider et al., mitotic activity correlated with prognosis (74). There were no relapses in 35 patients whose tumors exhibited low or moderate mitotic activity. Among those with high mitotic activity (>19 mitoses per 10 HPFs), approximately half recurred.
Serum Markers
Although information specific to juvenile granulosa cell tumors is limited, the various tumor markers discussed above for AGCTs would appear to be applicable to JGCTs for monitoring of recurrent disease.
TUMORS IN THE THECOMA-FIBROMA GROUP
Considering that the ovarian stromal cell is the precursor of both fibroblasts and theca cells, pure thecomas and pure fibromas appear to represent extremes in a continuum, with a significant percentage of the tumors having admixtures of lipid-laden, steroid-secreting cells and collagen-producing spindle cells. Nonetheless, the vast majority of tumors in the thecoma-fibroma group are readily subcategorized based on relatively distinct clinical and histologic characteristics. The major subcategories include thecoma, fibroma-fibrosarcoma, and the sclerosing stromal cell tumor (SST).
Thecoma
Theca cell tumors (TCTs), or thecomas (Fig. 26.4), are composed of lipid-laden stromal cells, occasionally demonstrating luteinization, and are almost invariably clinically benign (4,77,78). Thecomas account for approximately 1% of ovarian neoplasms and occur at a more advanced age than other SCSTs. The majority of patients are in their sixth and seventh decades at the time of diagnosis (4,77). Combining 2 large series totaling over 140 patients, less than 10% presented prior to age 30 years. Notably, the luteinized tumors are an exception to this generalization, with 30% occurring in women before their fourth decade of life (79). Assessing a compendium of nearly 300 cases, bilaterality occurs with a frequency of approximately 2% and extraovarian spread occurs rarely, if at all (4,77,78,80).
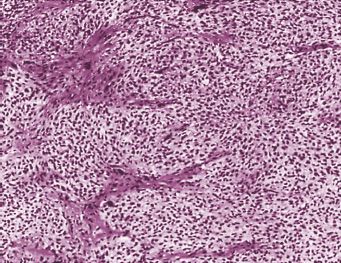
FIGURE 26.4. Thecoma. The tumor is composed of a mass of clear, vacuolated cells with round to oval nuclei intersected by bands of fibromatous tissue.
Source: Reprinted with permission from Morris JM, Scully RE. Endocrine Pathology of the Ovary. St. Louis, MO: Mosby; 1958.
The primary presenting signs and symptoms in patients with TCTs are abnormal vaginal bleeding and/or an abdominal/pelvic mass (77,78,80). The former urges initiation of medical intervention in the majority of postmenopausal patients, whereas an increasing abdominal girth or a palpable mass is more frequently the main presenting complaint of premenopausal patients. Lesion size has been reported to vary from less than 1 to 40 cm in diameter (77,78,80). Ascites is occasionally encountered.
Thecomas are considered to be among the most hormonally active of the SCSTs. The abnormal bleeding encountered in 60% of patients is presumably attributable to excess estrogen production (77,78). In the series reported by Evans et al. (4), endometrial hyperplasia was observed in 37% of the evaluable patients, and adenocarcinoma consistent with an unopposed estrogen effect was documented in an additional 27%. All the uterine cancers were well differentiated and minimally invasive, but 2 patients subsequently died of endometrial carcinoma. Other coexisting uterine pathologic findings potentially influenced by elevated circulating estrogen levels included leiomyomata, myohypertrophy, and endometrial polyps. Conversely, Zhang et al. (79) noted that nearly one-half of the evaluated luteinized thecomas were either nonfunctional or androgenic, resulting in a relatively significant frequency of masculinization.
An enigmatic tumor that has been considered to be a variant of luteinized thecoma has been associated with sclerosing peritonitis (81). These tumors are often bilateral and frequently have a brisk mitotic rate, but have not been shown to have metastatic potential. Sclerosing peritonitis has, however, been fatal owing to complications pursuant to it. Schonman et al. present a case of luteinized thecoma associated with sclerosing peritonitis treated with ovarian wedge resection (82). Despite a recurrent bowel obstruction, she was successfully treated with high-dose steroids. She achieved pregnancy and was free of symptoms 18 months following treatment.
Fibroma-Fibrosarcoma
Fibromas represent the most commonly encountered SCST, accounting for approximately 4% of all ovarian neoplasms. These endocrine-inert tumors are seldom bilateral and vary in size from microscopic to extremely large masses. Although infrequently diagnosed prior to age 30 years, fibromas can occur at any age; the average age of diagnosis is the latter half of the fifth decade of life (83). As their size increases, fibromas tend to become more edematous, leading to the escape of increasing quantities of fluid from the tumor surfaces. Ascites is detected in association with 10%–15% of ovarian fibromas exceeding a diameter of 10 cm (84). Furthermore, 1% of patients develop a hydrothorax in addition to the hydroperitoneum (Meigs’ syndrome) (85). Gorlin’s syndrome represents an inherited predisposition to the development of ovarian fibromas along with several other abnormalities, the most frequent of which is the appearance of basal-cell nevi at an early age (86).
Although ovarian fibromas are generally benign, approximately 10% will demonstrate increased cellularity and varying degrees of pleomorphorism and mitotic activity. Fibromatous tumors characterized histologically by an increased cellular density and brisk mitotic activity are designated cellular fibromas and are considered to be tumors of low malignant potential, particularly if ruptured or associated with adhesions (87). In contrast, fibrosarcomas are highly malignant neoplasms. These tumors are distinguished by their greater cellular density and, most notably, moderate to marked pleomorphorism (88).
Sclerosing Stromal Cell Tumors
SSTs were initially described by Chalvardjian and Scully (89) in 1973 as a distinct subgroup within the thecoma-fibroma family of ovarian tumors. Accounting for less than 5% of SCSTs, this relatively rare tumor characteristically differentiates itself histologically and clinically from both thecomas and fibromas (90,91). Histologically, the presence of pseudolobulation of cellular areas separated by edematous connective tissue, increased vascularity, and prominent areas of sclerosis are distinguishing features. Clinically, SSTs commonly become manifest during the second and third decades of life, with 80% being diagnosed prior to age 30 years, which is unique among ovarian stromal tumors (92). The signs and symptoms that most commonly necessitate medical evaluation include menstrual irregularities and/or pelvic pain (93). Despite the relatively large tumor size, which ranges from clinically undetectable to 20 cm or more in greatest diameter, ascites is seldom encountered; this further contrasts SSTs from fibromas (93). In contrast to thecomas, SSTs were originally considered to be inactive endocrinologically (89). However, a limited number of cases have been subsequently reported in which steroidogenic activity has been clinically demonstrable (93–96). To date, all SSTs have been clinically benign, and with one exception (97), all have been unilateral. Although a recent report noted an elevated CA-125 level, which the investigators speculated was perhaps nonspecific (98), no specific tumor marker has been identified for SSTs to date.
Natural History
Ovarian thecomas, fibromas, and SSTs are considered to be benign neoplasms, and any associated morbidity or mortality is attributable to the treatment modalities or the sequelae of concurrent disease (4,77,80,83,99). Examples of the latter are deaths from endometrial carcinoma resulting from the unopposed estrogen produced by the thecomas (4). Although several cases of “malignant thecomas” have been reported, critical reappraisal of such tumors invariably results in histologic reassignment to sarcomas or diffuse granulosa cell tumors (100). DNA ploidy analyses of thecomas and fibromas demonstrate aneuploid patterns in the majority, most commonly trisomy and/or tetrasomy 12, which offer a clue to their pathogenesis (101,102).
Prognostic Factors
The prognosis for patients diagnosed with cellular fibromas is generally considered to be quite favorable. Recurrences of these tumors of low malignant potential are generally correlated with adherent disease, rupture, or incomplete removal at the time of primary cytoreduction (87). Fibrosarcomas are associated with an extremely poor prognosis, but fortunately they are rare.
SERTOLI STROMAL CELL TUMORS
Neoplasms arising in the ovary exhibiting morphologic characteristics similar to those of the testes during various stages of gonadogenesis were recognized and elegantly described by Meyer (103,104). He reasoned that the origin of these tumors was the male blastema and coined the term arrhenoblastoma. Considering the functional nature of these male homologs and the varying degrees of associated defeminization and/or masculinization, the term androblastoma was also adopted. However, Morris and Scully (105), in 1958, contended that both designations implied masculinization, which is frequently absent, and furthermore facilitated the inclusion of a variety of unrelated androgen-producing ovarian tumors. Therefore, they recommended the adoption of the morphologic designation Sertoli-Leydig cell tumor (SLCT), which also allowed a consistent nomenclature for the general classification of SCSTs of the ovary. The SLCTs include tumors composed of Sertoli cells only, Leydig cells only, and a combination of Sertoli and Leydig cells.
Sertoli Cell Tumors
Sertoli cell tumors are rare, accounting for less than 5% of all SLCTs (92). The average age at presentation is about 30 years, but this lesion can occur at any age. Evidence of estrogen production has been observed in approximately two-thirds of the reported cases. Consistent with excess estrogen production, isosexual precocious puberty has been witnessed during the first decade of life, menstrual disorders during the reproductive decades, and postmenopausal bleeding in the decades after the climacteric. Reflecting tumor size (average 9 cm), capsular distention, and/or adnexal torsion, and abdominal distention and/or pain are frequent complaints. Pelvic examination generally confirms the presence of the tumor under these circumstances. The frequency with which excessive renin production has been associated with Sertoli cells appears to exceed mere chance (106–108). Evaluation of refractory hypertension and hypokalemia has rarely eventuated in the discovery of a Sertoli cell tumor as the origin of the excess renin. An occasional Sertoli cell tumor has arisen in a patient with the Peutz-Jeghers syndrome (PJS) (109).
Pathology
On gross examination, these rare tumors are typically solid, lobulated, and yellow (92,110). Microscopic examination typically shows hollow or solid tubules lined by cells that usually have relatively bland cytologic features, but rare tumors exhibit moderate to severe nuclear atypia. In most tumors, a tubular pattern predominates, but occasionally a diffuse pattern is conspicuous.
Prognosis
The great majority of these rare tumors have been unilateral stage I lesions. The greater majority of Sertoli cell tumors are well differentiated, and only rare tumors are malignant (92). Excision of the tumor results in prompt resolution of the hyperestrogenic state. (Leydig cell tumors are discussed in the section on steroid cell tumors below.)
Sertoli-Leydig Cell Tumors
SLCTs are extremely uncommon, accounting for less than 0.2% of all ovarian tumors. As implied by their designation, the tumors contain both Sertoli and Leydig cell elements. Many of the clinical characteristics are related to the degree of histologic differentiation and the presence of a retiform pattern and/or heterologous elements (described below). The average patient age at diagnosis is approximately 25 years, with the majority (70%–75%) of the tumors becoming clinically manifest during the second and third decades of life. Less than 10% occur either prior to menarche or after the climacterium. Patients harboring well-differentiated tumors present at an average age of 35 years, or 10 years later than patients with intermediate or poorly differentiated lesions. Conversely, tumors with retiform patterns are generally detected 10 years earlier than the intermediate and poorly differentiated tumors (111–113). Based on the compiled data from 3 reported series totaling over 300 patients, the frequency of extraovarian spread of disease at the time of diagnosis is approximately 2% to 3%. In addition, the likelihood of encountering bilateral tumors is even less frequent (111–113).
The most frequent complaints at the time of presentation of these generally healthy adolescents and young adults are menstrual disorders, virilization, and nonspecific symptoms resulting from an abdominal mass. Nearly one-half of the patients experience sufficient abdominal pain or discomfort or note abdominal distention or palpate a mass on self-examination to prompt professional assessment. Whereas capsular distention and/or intralesional hemorrhage or necrosis of the tumor and/or adjacent visceral compression by the tumor account for the associated chronic or intermittent pain, acute abdominal pain necessitating emergency intervention invariably reflects vascular compromise from torsion. While lesion size varies according to histologic differentiation (approximately 5 cm for well-differentiated tumors to >15 cm for poorly differentiated tumors), abdominal, vaginal, and/or rectal examination readily identifies an adnexal mass in approximately 95% of symptomatic patients. The most common premonitory symptoms, namely, menstrual disorders and subtle androgenic manifestations, predate by several months, and less often, by years, the recognition of the overt clinical signs or symptoms. Irregular bleeding, oligomenorrhea, and postmenopausal bleeding, retrospectively, have been attributed to either excess androgens or estrogens. The etiology of the latter is presumably the peripheral conversion of androgens to estrogens or, rarely, from an estrogen-secreting SLCT. Frank virilization occurs in 35% of the patients with SLCTs, and another 10% to 15% have some clinical manifestations consistent with androgen excess. The most frequent androgenic symptom complex encountered includes amenorrhea, voice deepening, and hirsutism. In addition, breast atrophy, clitorimegaly, loss of female contour, and temporal hair recession, for example, are signs of masculinization witnessed in patients with SLCTs (111–113). The prevalence of androgenic manifestations appears to be independent of the degree of histologic differentiation, but is observed less frequently in heterologous SLCTs and only occasionally in patients harboring retiform lesions (112,114–117). Although the preoperative diagnosis of SLCT in the absence of androgenic excess may be impossible, this neoplastic entity should constitute the primary preoperative diagnosis in patients with androgenic manifestations presenting during the second through the fourth decades of life with a unilaterally palpable adnexal mass.
Uncommonly, estrogen manifestations are witnessed in the context of presumed end-organ estrogenic responses, including postmenopausal bleeding and endometrial polyp formation, hyperplasia, and adenocarcinoma. Cautious interpretation of such observations is required, realizing that peripheral conversion of androgens to estrogens may be as plausible as a primary estrogen-secreting SLCT. As expected from the clinical findings, most patients demonstrating signs of defeminization or virilization have elevated plasma testosterone levels (111–113). Whereas plasma androstenedione may occasionally be elevated, the urinary 17-ketosteroids, including dehydroepiandrosterone, are usually normal, with the occasional patient presenting with a slightly elevated level. An elevated testosterone/androstenedione ratio generally suggests the presence of an androgen-secreting ovarian tumor, most likely an SLCT. Recognizing that certain gonadotropin-releasing hormone (GnRH) agonists modulate androgen production by downregulating gonadotropin levels and through a direct effect on the ovary, Pascale et al. (118) demonstrated successful suppression of testosterone and androstenedione in five virilized women with the administration of GnRH agonists. Their data suggest that androgen-secreting tumors of the ovary appear to be less autonomous than such tumors originating in the adrenal gland. Surgical excision of the SLCTs results in a precipitous drop in androgen levels and, over time, partial to complete resolution of the clinical manifestations associated with androgen excess is observed.
The coexistence of other diseases with SLCTs has been chronicled. The frequency with which thyroid disease is observed in these patients appears to exceed mere chance. Furthermore, several cases of other mesenchymal tumors have occurred in patients with SLCTs, including sarcoma botryoides of the cervix as well as Ollier’s disease (112,119). The latter is a rare disease, but it is associated with other SCSTs, specifically JGCTs, as noted above. Finally, a tendency toward familial occurrence appears to exist (112).
Pathology
Gross Features
Sertoli-Leydig cell tumors vary in size from small to huge masses, but most are between 5 and 15 cm in diameter. The majority are solid, often yellow, and lobulated (Fig. 26.5), but many are solid and cystic. Pure cystic tumors are exceptionally rare, in contrast to the situation with GCTs. Poorly differentiated tumors tend to be larger than those more differentiated, and contain areas of hemorrhage and necrosis more frequently (112). Tumors with heterologous or retiform components are more often cystic than other tumors in this category (114,115,117,120). The heterologous tumors occasionally simulate mucinous cystic tumors on gross examination, and retiform tumors may contain large edematous intracystic papillae, resembling serous papillary tumors, or may be soft and spongy with varying degrees of cystification (117).
Microscopic Features
Well-differentiated SLCTs are characterized by a predominantly tubular pattern (121). On low-power examination, a nodular architecture is often conspicuous, with fibrous bands intersecting lobules composed of small, round, hollow, or, less often, solid tubules lined by well-differentiated cells and separated by variable numbers of Leydig cells. Rarely the tubules appear pseudoendometrioid (122).
Sertoli-Leydig cell tumors of intermediate and poor differentiation form a continuum characterized by a variety of patterns and combinations of cell types (111–113). Some tumors exhibit intermediate differentiation in some areas and poor differentiation in others, and less commonly, tumors of intermediate differentiation contain well-differentiated foci. Both the Sertoli cells and the Leydig cells may exhibit varying degrees of immaturity. In tumors of intermediate differentiation, immature Sertoli cells have small, round, oval, or angular nuclei and generally scanty cytoplasm and are arranged typically in ill-defined masses, often creating a lobulated appearance on low power; solid and hollow tubules, nests, broad columns of Sertoli cells, and, most characteristically, thin cords resembling the sex cords of the embryonic testis are often present (Fig. 26.6). These structures are separated by stroma, which ranges from fibromatous to densely cellular to edematous, and typically contains clusters of well-differentiated Leydig cells (Fig. 26.6). Cysts containing eosinophilic secretion may be present and create a thyroid-like appearance, and follicle-like spaces are encountered rarely. The Sertoli cell and Leydig cell elements, singly or in combination, may contain varying and sometimes large amounts of lipid in the form of small or large droplets. When a significant amount of the stromal component is made up of immature cellular mesenchymal tissue with high mitotic activity resembling a nonspecific sarcoma, the tumor is poorly differentiated.
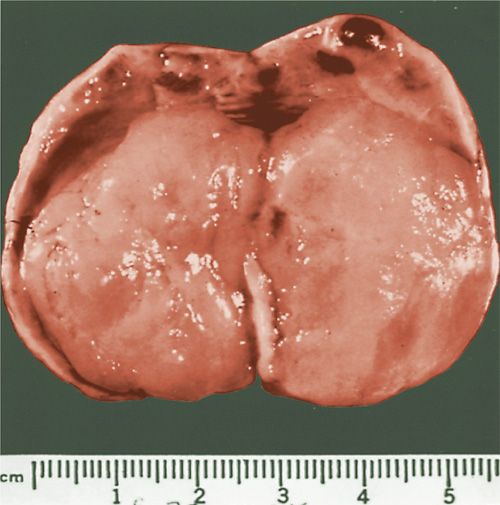
FIGURE 26.5. Sertoli-Leydig cell tumor. The sectioned surface of the tumor is focally lobulated and was yellow in the fresh state.
Fifteen percent of SLCTs have a substantial retiform component, and are so designated because they are composed of a network of elongated tubules and cysts, both of which may contain papillae, resembling the rete testis (32). This pattern is usually accompanied by other patterns of SLCTs, but sometimes an entire tumor has a retiform pattern.
Heterologous elements occur in approximately 20% of Sertoli cell tumors (118,123). In a series of these tumors, 18% contain glands and cysts lined by moderately to well-differentiated intestinal-type epithelium (118). Mesenchymal heterologous elements, encountered in 5% of Sertoli cell tumors, include islands of cartilage arising on a sarcomatous background, areas of embryonal rhabdomyosarcoma, or both (123).
Natural History
SLCTs display characteristics that differ markedly from their epithelial counterparts, notably in regard to their malignant potential. Despite an average size of approximately 16 cm, only 2% to 3% of SLCTs have demonstrable extraovarian spread at the time of detection. Furthermore, Young et al. (62) identified only 29 clinically malignant cases in their series of 220 SLCTs having variable observation intervals, and they noted an 18% malignancy rate among 164 patients with adequate follow-up. At least in part, the more favorable prognosis reflects the abrupt onset of androgenic manifestations and the early detection of nonspecific symptoms, which promote prompt medical assessment. Nevertheless, the natural history of the malignant variant includes early recurrences, with approximately two-thirds becoming evident within 1 year of treatment and only 6%–7% recurring after 5 years. The abdominal cavity (including the pelvis) and the retroperitoneal nodes are the most frequent sites for recurrences. In addition, the contralateral ovary, lungs, liver, and bone are other reported sites of recurrent metastatic disease. The collective salvage rates in patients with clinically malignant disease are low, with extrapolated estimates being less than 20%.
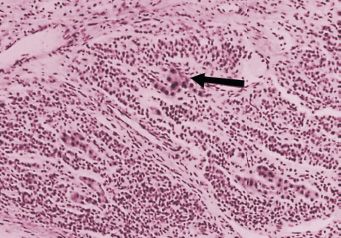
FIGURE 26.6. Sertoli-Leydig cell tumor, intermediate differentiation. Nests of large Leydig cells (arrow) lie among bands of immature Sertoli cells.
Source: Reprinted with permission from Morris JM, Scully RE. Endocrine Pathology of the Ovary. St. Louis, MO: Mosby; 1958.
Prognostic Factors
Stage is the most important predictor of outcome in SLCTs. Fortunately, 97% of SLCTs are reportedly stage I at diagnosis, and less than 20% of these localized tumors become clinically malignant. The most cogent phenotypic prognostic determinant for stage I SLCTs is the degree of histologic differentiation (111–113
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


