Oncology
Patrick T. McGann
Kevin R. Schwartz
Howard J. Weinstein
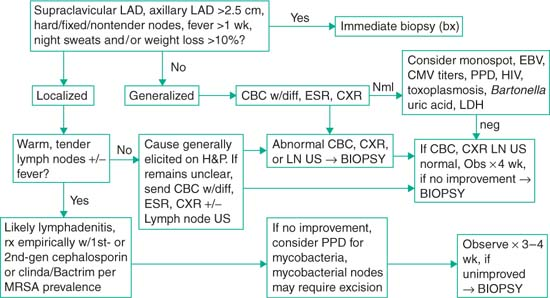
Lymphadenopathy in the Pediatric Patient
Definition
(Pediatr Rev 2008;29:53)
Axillary or cervical lymph node >1 cm in size; inguinal lymph node >1.5 cm; epitrochlear lymph node >0.5 cm; supraclavicular—any palpable lymph node
Note: Risk of underlying malignancy increases w/ increasing size, esp >2 cm
Localized lymphadenopathy: Involving one nodal group
Generalized lymphadenopathy: Involving ≥2 nodal groups or sites (i.e., spleen/liver)
Differential Diagnosis of Lymphadenopathy
(Pediatr Rev 2008;29:53; Pediatr Rev 2000;21:399)
Infections
|
| Malignancy: Leukemia, lymphoma, metastasis from solid tumor Immunologic: Angioimmunoblastic LAD w/dysproteinemia, autoimmune lympho-proliferative dz, chronic granulomatous dz, dermatomyositis, drug rxn, RA, hemophagocytic lymphohistiocytosis, Langerhans cell histiocytosis serum sickness, SLE Endocrine: Addison disease, hypothyroidism Misc: Amyloidosis, Castleman dz, Churg-Strauss, inflammatory pseudotumor Kawasaki dz, Kikuchi dz, lipid storage dz Sarcoidosis |
Clinical Manifestations
(Pediatr Clin North Am 2002;49:1009)
Hx: Duration LAD, assoc sx, recent localized infxn’s (esp in drainage territory of nodes), skin lesions, trauma, animal scratches/bites, meds, ingestion of unpasteurized milk/undercooked meats, dental problems (cervical LAD), tick bites, travel Hx, sexual Hx.
Exam: Size, location (supraclavicular always concerning), tender/nontender, mobile/fixed, soft/hard, warm or erythematous, HSM, bruises or petechiae, signs of systemic dz.
Pediatric Oncologic Emergencies
(Principles & practice of pediatric oncology. 5th ed. Philadelphia. LWW 2006;12224)
Superior Vena Cava (SVC) Syndrome
(Pediatr Clin North Am 1997;44:809)
Definition: Signs and sx’s from compression &/or obstruction of SVC
Etiology: Often a presenting feature of intrathoracic malignancies
Malignancies p/w mediastinal mass most commonly include NHL, ALL, and Hodgkin dz. Less common: Sarcoma (Ewing, rhabdo), neuroblastoma, thymoma, and teratoma.
Pathophysiology: Mediastinal mass compresses SVC causing venous stasis
Compression, clotting, and edema of SVC lead to ↓ in tracheal airflow, and ↓venous return from head, neck, and upper thorax
Clinical manifestations: Cough, dyspnea, dysphagia, orthopnea, and hoarseness
Later sx’s of anxiety, confusion, lethargy, HA, Δ vision, and syncope may = CO2 retention; can also see facial +/- UE swelling, chest pain, pleural effusions
Sx’s worse when patient is supine; should raise suspicion for mediastinal mass
Diagnostic studies: CXR; typically will show anter mediastinal mass; trach deviation
CBC, Chem10, LDH, and uric acid; Obtain dx tissue sample before Rx if possible
Treatment: Depends on underlying malignancy
If significant CV or resp compromise, emergent XRT and/or IV methylprednisolone/dexamethasone treatment may be indicated
Tumor Lysis Syndrome (TLS)
(Nat Clin Pract Oncol 2006;3:438)
Definition: Metabolic abn 2/2 cell death and subsequent release cell contents into circ.
Metabolic disturbances can result in severe end organ impairment
Etiology/risk factors
Occurs in tumors w/ ↑ growth fraction and large tumor burden/volume.
Malignancies most commonly assoc w/ TLS include Burkitt lymphoma, ALL (particularly T-cell variant), lymphoblastic lymphoma; TLS rare in AML
Can occur before onset of Rx but typically occurs w/i 12–72 hr of initiation of Rx
Risk factors: WBC >50,000, ↑ LDH, ↑ uric acid on admit, Cr >1.6 or ↓ GFR
Diagnostic studies: Elevated uric acid (>10) – caused by breakdown of nucleic acids
Hyperphosphatemia and secondary hypocalcemia; hyperkalemia
Consequences of TLS
Hyperuricemia → precipitation of uric acid in collecting ducts of renal tubules, causing resultant nephropathy and acute renal failure
Hyperkalemia → cardiac arrhythmias and sudden death
Hypocalcemia → hypotension, EKG changes, tetany, and seizures
Hyperphosphatemia → renal precipitation; exacerbate nephropathy and renal failure
Management/prevention of TLS: Electrolyte abn managed acutely as indicated
Upon dx of malignancy and before starting Rx, aggressive mgmt to prevent TLS
Aggressive hydration (2–4× maintenance) to ↑ GFR and ↑ urinary outflow
Urinary alkalization w/ NaHCO3(40–80 mEq/m2/hr) to urine pH >7 to prevent uric acid precipitation
Allopurinol (250–500 mg/m2/d) inhibits xanthine oxidase; ↓ uric acid formation
Alternatively, rasburicase (recombinant urate oxidase) in place of allopurinol
Close observation of electrolytes, Ca, Mg, Ph, LDH, and uric acid (q6–8h upon initiation of Rx) essential to monitor for development of TLS
Spinal Cord Compression (SCC)
(Pediatr Clin North Am 1997;44:809)
Definition/etiology: Occurs in 2.7%–5% of children w/ cancer
Most cases 2/2 epidural compression from extension of paravertebral tumor
↑ risk w/ neuroblastoma, Ewing sarcoma, non-Hodgkin lymphoma, and Hodgkin.
Osteosarcoma and rhabdomyosarcoma typically cause SCC only w/ recurrence
Clinical manifestations: Back pain present in 80% pedi pts w/ cord compression
Sx’s typically present for an avg of 2 wk before dx made
Weakness, sensory loss, and incontinence are later and more concerning findings
Evaluation/treatment
Detailed neuro exam in any pt p/w suspected malign; rectal exam for sphincter tone
Plain radiographs often performed but only show findings in ½ affected patients
MRI w/ contrast is study of choice to assess presence and extent of SCC
Children w/ neuro findings and/or rapidly progression spinal cord dysfxn should receive dexamethasone 1 mg/kg IV and have emergent spinal MRI
Children w/o neuro sx’s →MRI w/i 24 hr; low-dose dexamethasone PO (0.25–0.5 mg/kg)
Surgery, XRT, and chemo are other emergent Rx options depending on tumor type
Hyperleukocytosis
(Pediatr Clin North Am 1997;44:809)
Definition/etiology: WBC >100,000; Presence high # of circ leukemic blast cells
Clinically signif hyperleukocytosis >200,000 in AML, >300,000 in ALL and CML
Occurs in 9%–13% of patients with ALL and 5%–22% of patients with AML
Pathogenesis: Excessive leukocytes obstruct circulation in brain, lung, and other organs forming aggregates and white thrombi in small veins
Excessive leukocytes also compete for oxygen and damage vessel walls
Morbidity is directly related to blood viscosity
Myeloblasts and monoblasts are larger and more likely to cause obstruction
Clinical manifestations
Pulmonary leukostasis→ dyspnea, hypoxia, and right ventricular failure
Intracerebral leukostasis→ Δ mental status, frontal HA, szr’s, and papilledema
Other possible complications include priapism, renal failure, and dactylitis
Major complications of hyperleukocytosis in ALL usually result of TLS
Complications of hyperleukocytosis in AML usually are result of intracerebral leukostasis and include stroke and hemorrhage
Treatment: Aggressive hydration, alkalization, and allopurinol to minimize risk of TLS
Maintain platelet count >20,000 because of risk of intracranial hemorrhage
Maintain Hgb level <10 and minimize RBC xfusion to prevent further ↑ in viscosity
Correct coagulopathy w/ FFP and vitamin K as indicated
Exchange xfusion and leukophoresis also used to help rapidly ↓ leukocyte count
Neutropenia in the Pediatric Patient
(Pediatr Rev 2008;29:12)
Definition
Absolute neutrophil count (ANC) = WBC (cells/μL) × %(PMNs + bands) ÷ 100
Mild Neutropenia: ANC 1000–1500/μL; moderate neutropenia: ANC 500–1000/μL; severe neutropenia: ANC <500/μL
Note: Lower limit nml ANC = 1500 for pts >1 yo. For pts 2 wk–6 mo, nml ANC >1000. Pts <2 wk nml ANC variable. 3%–5% of African Americans have ANCs <1500 normally.
Differential Diagnosis
(Pediatr Rev 2008;29:12)
Acquired causes:
Infection:
Viral (most common cause neutropenia, 2/2 BM suppression, lasts 3–8 d): EBV, CMV, parvo, RSV, flu A/B, hepatitis, HHV6, VZV, rubella, rubeola, HIV
Bacterial: Typhoid fever, Shigella, brucellosis, tularemia, TB, malaria, RMSF
Drug-induced: PCNs, sulfonamides, chloramphenicol, phenytoin, ibuprofen, ranitidine, hydralazine, carbamazepine, cimetidine, chlorpromazine, indomethacin, quinidine, propylthiouracil, procainamide, chlorpropamide, phenothiazines.
Immune:
Neonatal alloimmune neutropenia (maternal antineutrophil IgG xfer across placenta, dx. w/ antineutrophil Ab in infant and maternal serum; self-resolves by 2–3 mo)
Primary autoimmune neutropenia (neutropenia may be severe, typically presents btw 5–15 mo, dx by detecting antineutrophil Ab in pt.)
Secondary autoimmune neutropenia (e.g., SLE, Evans syndrome)
Sequestration (typically mild neutropenia w/ splenomegaly of any cause)
Nutritional def (typically p/w anemia, hypersegmented PMNs): B12 or folate def
Chronic idiopathic
Inherited causes:
Severe congenital neutropenia (early infancy w/ severe neutropenia and infxns, may be AR = Kostmann syndrome (HAX1 gene mut) or AD (ELA2 and GFII mut’s. High risk of later developing MDS and/or AML)
Cyclic neutropenia (characterized by 21-d cycles w/ neutropenia lasting 3–6 d in a cycle, sometimes severe. Dx w/ serial CBCs 2–3×/wk × 4–6 wk.)
Shwachman-Diamond syndrome (mild-mod neutropenia + exocrine pancreatic insuff, short stature, metaphyseal dysplasia. ↑ risk for MDS/AML)
Marrow failure syndromes: Fanconi anemia(pancytopenia usually in 5–10 yo), dyskeratosis congenita (neutropenia, abn skin pigmentation, dystrophic nails, leukoplakia), Diamond-Blackfan syndrome (anemia, thumb, and craniofacial anom)
Syndromes w/ associated immunodeficiencies:
Hyper-IgM syndrome, dys-γ-globulinemia, myelokathexis, Chediak-Higashi (albinism, perph neuropathies), cartilage hair syndrome (fine hair, short-limbed, dwarfism, lymphopenia), Wiskott-Aldrich (eczema, neutropenia, thrombocytopenia), selective IgA def, reticular dysgenesis (SCID w/ neutropenia)
Initial Evaluation
(Pediatr Rev 2008;29:12)
Hx: Hx of infxn’s (esp mouth ulceration), cong anomalies, med exposures, recent illnesses. FHx for neutropenias or serious infxn’s, hospitalizations, blood dz’s
Exam: Physical feature c/w immunodeficiency syndrome (see earlier discussion).
Laboratory eval: CBC w/ diff & retic count. Consider rpt CBC to verify.
Further eval: Per suspected cause: observation (if mild/mod neutropenia and suspected viral cause, rpt CBC 1–2 wk), viral titers/tests, antineutrophil Ab (if AI suspected), serial CBCs (if cyclic suspected), DNA analysis for HAX1, ELA2, GFII (if severe congenital suspected), quant Igs w/ B and T cell subsets (if evidence of immunodef), ANA/anti-dsDNA (if SLE suspected), BM biopsy/aspirate (if multiple lines affected or dx unclear)
Acute Lymphoblastic Leukemia
Definition:
Clonal proliferation of either pre-B, mature B or T lymphocyte cell lines.
Epidemiology
(Pediatr Rev 2005;26:96)
∼2500–3500 new pedi cases/yr; most common pedi cancer (25% all pedi cancer)
May occur at any age with peak incidence 2–5 yo; Males > Females, whites > blacks.
Risk factors: Down syndrome, neurofibromatosis type 1, ataxia telangiectasia, Fanconi, other syndromes.
Clinical Presentation
(Pediatr Rev 2005;26:96)
Fever (55%), bleeding +/- petechiae/purpura (45%), malaise (40%), bone pain (30%), hepatomegaly (70%), splenomegaly (50%), LAD (50%), abd pain (10%).
CBC: Leukocytosis or leukopenia and anemia (88%) and/or thrombocytopenia (80%)
Blasts may be visible on peripheral blood smear, but are not always present.
Prognostic Factors and Overall Prognosis
(N Engl J Med 2006;354:166)
Overall survival: ∼80 %
Favorable prognostic features: Age at presentation 1–9 yo, presenting WBC <50,000; favorable cytogenetics include: Hyperdiploidy (>50 chromo) w/ trisomies of 4, 10, and 17, t(12;21)/TEL-AML1 fusion gene.
Unfavorable prognostic features: Age <1 yo, presenting WBC >50,000, highly unfavorable cytogenetics: Hypoploidy (33–39 chromo) or near haploploidy (23 to 29 chromosomes), t(4;11)/MLL-AF4 fusion gene in infants (present in 80% of infant ALL), and t(9;22)/BCR-ABL protein/Philadelphia chromosome.
Other unfavorable features; fail to achieve morph remission after induction or >1% blasts detectable by PCR or flow cytometry (min residual dz) at the end of induction
Treatment
(Pediatr Clin North Am 2008;55:1; N Engl J Med 2006;354:166)
Total treatment duration is typically 2–3 yr and consists of 3 phases:
Induction (4–6 wk): Prednisone or dexamethasone, vincristine, PEG L-Asparaginase +/- anthracycline, also w/ intrathecal Rx (d 1 and 8 for CNS)
Consolidation (4–6 mo): High-dose methotrexate, cyclophosphamide, cytosine arabinoside (ara-C), 6-MP, and PEG L-asparaginase.
Most protocols have delayed intensification or reinduction phase; 4–6 wk pulse intensive Rx similar/identical to induction during 1st 6 mo of remission.
Maintenance (1.5–2.5 yr): Methotrexate weekly and mercaptopurine daily
Role of BMT: Reserved for pts at very high risk; those w/ t(9;22)/Philadelphia chromosome or those with poor initial response to treatment.
Role of XRT: Cranial radiation therapy reserved only for very high-risk patients or those with significant CNS leukemic involvement
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


