Nonsteroidal Anti-Inflammatory Drugs
Sylvain Chemtob
Véronique G. Dorval
Inflammation is among the most important tissue responses to injury; it is observed in all acquired disorders as well as genetic conditions modified by environmental exposures. The outcome of the inflammatory response may be beneficial as is the case in limiting invading organisms. On the other hand, the outcome may be deleterious if chronic inflammation ensues, as seen with chronic rheumatic disorders, which lead to pain and destruction of bone and cartilage resulting in potentially severe disability. Inflammation involves a complex sequence paradigm implicating responses of a number of cell types, such that upon early activation of endothelium, leukocytes and monocytes are chemoattracted to the site to contain the injury. The elimination of triggering noxious stimulus is associated with sacrifice of certain cells. Repair involves repopulation of some cells associated with revascularization.
There are numerous mediators of inflammation and their properties are intertwined with one another. For instance, effects of chemokines, interleukins, leukotrienes, and histamine are interlinked in lung inflammation associated with asthma; while in degenerative arthritis interleukins (especially IL-1 and IL-18) interact with prostanoids. Growth factors such as platelet-derived growth factor and vascular endothelial growth factor participate in these processes, especially during the repair phases. In this chapter, we shall focus on lipid mediators modulated by nonsteroidal anti-inflammatory drugs (NSAIDs); we will also cover some pharmacology of acetaminophen which, although not classically an NSAID, may exert its effects (at least in part) via cyclooxygenases (COXs). A review of prostanoid biochemistry and pharmacology will be presented relevant to the mode of action of NSAIDs. Applications of NSAIDs particularly in pyrosis, arthritic conditions, and in indications applicable to the neonate, notably patent ductus arteriosus (PDA) and intraventricular cerebral hemorrhage, will be covered. Detailed descriptions of other agents indicated in pyresis, pain, asthma, some of which also act via prostanoids, will be covered elsewhere in this book.
Synthesis of Prostanoids
Prostanoids are important autacoids which exert diverse physiological and pathophysiological effects in various systems. These involve modulation of neuronal activity, pyrexia and sleep induction, alterations in platelet aggregation, relaxation and contraction of smooth muscle, regulation of ion and water transport in kidneys as well as of gastrointestinal motility and secretion. There are five physiologically major prostanoids that are well characterized, prostaglandin E2 (PGE2), PGF2 a, PGD2, PGI2, and thromboxane A2 (TXA2); other prostanoid-like compounds, such as isoprostanes, also exert biological effects through receptor sites that are not yet clearly identified (1,2,3), and these will not be covered in this review.
Prostanoids derive from arachidonic acid released from phospholipids. Cyclooxygenase (COX) is the enzyme responsible for the committed step in the conversion of arachidonic acid to prostanoids (Fig. 48.1). There are two separate genes encoding COX proteins, COX-1 and COX-2; a splice variant of COX-1, termed COX-3, has also been unidentified (4). COX-1 is mostly constitutive, and COX-2 is highly and readily expressed during inflammation. However, COX-1 also contributes significantly to prostaglandin generation in inflamed tissue (5,6). Along the same lines, COX-2 is normally expressed in a number of tissues independent of inflammatory stimuli, especially during development. For instance, COX-2 is highly expressed in kidneys, and disruption of its gene (in mice) leads to fatal renal failure (7,8); adverse renal effects have also been observed with COX-2 inhibitors (9). COX-2 is also normally expressed in perinatal brain (10).
Both COX-1 and COX-2 catalyze the same reactions by converting arachidonic acid into PGH2. PGH2 is then converted into specific prostanoids by individual enzymes. PGE2 formation is catalyzed by at least one cytosolic and two microsomal isoforms (11,12,13); PGD2 by three PGD2 synthases of which the principal one is lipocalin PGD2 synthase; and PGF2 a, PGI2, and TXA2 by specific individual
enzymes; synthases for the latter two exhibit cytochrome P450 properties, and belong to corresponding CYP8 and CYP5 subfamilies, respectively.
enzymes; synthases for the latter two exhibit cytochrome P450 properties, and belong to corresponding CYP8 and CYP5 subfamilies, respectively.
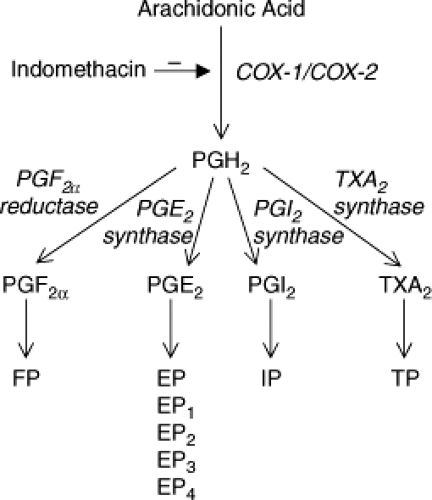 Figure 48.1. Arachidonic acid metabolism via the cyclooxygenase pathway. Nonsteroidal anti-inflammatory drugs (NSAIDs), such as indomethacin, inhibit cyclooxygenase (COX) conversion of arachidonic acid into prostaglandin H2 (PGH2). |
COX-3 [and partial COX-1 proteins (a and b)] is derived from the COX-1 gene. COX-3 retains intron 1 of the COX-1 gene, while partial COX-1 protein does not contain a core central sequence of COX-1 (amino acids 119 to 337) (4). These proteins are capable of catalyzing prostaglandin formation and seem to be expressed in brain and heart. Bioactivity of COX-3 revealed that it is inhibited by acetaminophen and phenacetin (acetaminophen prodrug) at IC50 of 100 to 500 μM. However, all NSAIDs tested were found to be inhibitors of COX-3 with even greater potency than acetaminophen and phenacetin, especially for diclofenac which had an IC50 of 8 nM compared with 35 to 40 nM for COX-1 and COX-2; more recent studies reveal interesting observations pointing to actions of acetaminophen primarily through COX-2 including in humans (14,15). Thus, although COX-3 may be a target for acetaminophen, its relevance regarding acetaminophen effects in the clinical setting is questionable.
Prostanoid Receptors
Prostanoids exert their effects through respective receptors. On the basis of pharmacological characterization, which was confirmed biochemically, distinct receptors have been identified for prostanoids. A classification for prostanoid receptors was proposed and adopted such that for PGD2, PGE2, PGF2 a, PGI2, and TXA2, they are classified, respectively, as DP, EP, FP, IP, and TP; EP receptors are further divided into EP1, EP2, EP3, and EP4 (16,17). The eight known types of prostanoid receptors are each encoded by an individual gene. The cloning of the prostanoid receptors identified another degree of heterogeneity secondary to mRNA splicing. Splice variants have been identified for the EP1, EP3, FP, and TP receptor homologues of various species. There are currently nine known subtypes of the human EP3 receptor: EP3-1a, EP3-1b, EP3-II, EP3-III, EP3-IV, EP3-V, EP3-VI, EP3-e, and EP3-f (18,19,20); subtypes of the EP3 receptors are distinguished by variances in the tail of the carboxy-terminal portion. Human subtypes also exist for the TP receptor (21), specifically TPa and TPb, which as for EP3 subtypes vary by the tail of their carboxy-terminal end. Although splice variants have been identified for the rat EP1 (22) and the ovine FP (23) receptors, which also differ by their carboxyl-terminal portions, human homologues to EP1 and FP subtypes have not been detected.
Sequence homology is mainly based on signaling pathway than on preferential ligand (24,25,26). The effects of prostanoid receptors on smooth muscle reflect this relationship. Thus, EP2, EP4, DP, and IP induce smooth muscle relaxation and are more closely related to each other than to the other prostanoid receptors. Similarly, EP1, FP, and TP receptors cause smooth muscle contraction and form another group based on sequence homology.
Prostanoid receptors are rhodopsin-type containing seven transmembrane domains, an extracellular amino- and intracellular carboxy-terminus, and belong to the superfamily of G protein-coupled receptors. There are 28 amino acid residues conserved within all prostanoid receptor sequences, and 8 of these are shared with other G protein-coupled receptors. These conserved regions are thought to play fundamental roles in the structure of the prostanoid binding domains. These residues are believed to be particularly important in receptor structure and/or function. For instance, an arginine in the seventh transmembrane domain conserved between all prostanoid receptors was proposed to be the binding site for the carboxyl moiety of the prostanoids (27,28); the conserved motif in the second extracellular loop may also function in this regard (27). In addition, glycosylation is required for ligand binding of certain prostanoid receptors (29).
Three clusters of related receptors have been defined based on their signaling: (i) DP, IP, EP2, and EP4; (ii) EP1, FP, and TP; and (iii) EP3. Prostanoid receptors in group (i) are linked to heterotrimeric G proteins that are composed of a Ga subunit that generally stimulates adenylate cyclase (designated Ga s) to produce cyclic AMP. Accordingly, its dominant effect on smooth muscle is relaxation. Prostanoid receptors in group (ii) couple mostly to increases in intracellular free Ca2+ through the activation by Ga q of phospholipase C, with subsequent inositol phosphate liberation. In smooth muscle, stimulation of this group of receptors leads to contraction. Group (iii), which constitutes the EP3 subtypes of the prostanoid receptor family, employs as its primary effector pathway inhibition of adenylate cyclase through the Ga i family (30). However, because of the variant molecular nature of EP3, the latter can couple to a variety of G proteins.
The effects of PGE2 and PGD2 in inflammation are well established (31). PGE2 through its EP3 receptor is also significantly implicated in pyrexia (32), whereas PGD2 via its DP receptor seems to contribute to allergen-evoked bronchoconstriction (33); moreover, the PGD2 metabolite 15-deoxy-Δ-PGJ2 (12,14) is a potent immune modulator.
Nonsteroidal Anti-Inflammatory Drugs
NSAIDs are a heterogeneous group of agents, which inhibit COX activity (Fig. 48.1). NSAIDs can be classified
based on chemical structure (Table 48.1). More recently, specific COX-2 inhibitors have been developed, and older NSAIDs found to exhibit COX-2-preferential properties; the latter has been the case for nimesulide, meloxicam, and etodolac (34).
based on chemical structure (Table 48.1). More recently, specific COX-2 inhibitors have been developed, and older NSAIDs found to exhibit COX-2-preferential properties; the latter has been the case for nimesulide, meloxicam, and etodolac (34).
Table 48.1 Classification of Nonsteroidal Anti-Inflammatory Drugs | ||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
NSAIDs exert antipyretic, anti-inflammatory, and antinociceptive effects. Despite the pleiotropic nature of prostaglandins in various physiological and pathological processes, the effects of NSAIDs cannot be solely attributed to inhibition of COX. Indeed, COX inhibitors exert a number of COX-independent actions. For instance, doses of aspirin needed to treat chronic inflammatory conditions are much higher than those required to inhibit prostaglandin synthesis. A number of cellular mechanisms affected by NSAIDs, such as angiogenesis, apoptosis, and cell cycle progression, have been observed at NSAID concentrations 100- to 1,000-fold greater than those needed to inhibit prostaglandin formation. In this process, a number of targets other than COX have been identified for NSAIDs, including nuclear factor κB (NF-κB), mitogen-activated protein kinase (Erk2), ribosomal S6 kinase 2, signal transducer and activator of transcription-1 (STAT1), peroxisome proliferator-activated receptor #979; (PPAR#979;), and Akt protein kinase (35,36,37,38). However, the effects of NSAIDs on these various targets differ between NSAIDs (39). Moreover, although effects on COX are relatively restricted to the S-stereoisomers, R-stereoisomers act on some of the targets mentioned above.
Nonetheless, a major mechanism of action of NSAIDs is through inhibition of prostaglandin biosynthesis. This is consistent with proinflammatory of certain prostaglandins (34), the fever-inducing actions of PGE2 via its EP3 receptors, and nociceptive actions of PGI2. The mechanism of action of acetaminophen is still debatable. Although attempts to explain its main mechanism of action in vivo through the inhibition of COX-3 have lately mostly been rejected (40,41), recent studies point to significant COX-2 inhibition (14) particularly in the physiological setting when arachinodic acid levels and peroxide tone are relatively low (42,43). Although, its primary site of action appears to be inhibition of PG synthesis, nociceptive modulation by acetaminophen may also arise from activation of descending serotonergic pathways (44,45). Elucidating these mechanisms at the molecular level is currently ongoing.
Pharmacokinetic Properties of NSAIDs
A detailed review on pharmacokinetics of NSAIDs in children is reported (46). Pharmacokinetics in children is notable by interindividual differences. In general, NSAIDs are rapidly absorbed from the intestine to reach maximal plasma concentrations within 1 to 2 hours. They tend to distribute in a small volume of distribution and are highly bound to plasma proteins; but in children, the apparent volume of distribution is greater than that in adults as shown for diclofenac, ibuprofen, ketorolac, and nimesulide for reasons that are not yet clear. Assuming, a comparable concentration–efficacy relationship, children may require higher loading doses. NSAIDs are metabolized via phase I and II biotransformations. Conjugation occurs mostly with glucuronic acid and sulfate producing mostly inactive compounds. Approximately 60% to 70% of metabolized agents are excreted in urine and the rest in feces.
Aspirin is rapidly absorbed enterally with a tmax of 2 hours. It is readily deacetylated by intestinal, hepatic, and blood esterases. Aspirin is metabolized by liver enzymes mostly into salicyluric acid, salicyl phenolic glucuronide, and salicyl acyl glucuronide and altogether account for 90% of metabolites which are inactive and eliminated by the kidneys; because of the ionized form of this acid, its renal elimination is favored in alkaline urine. Interestingly, aspirin metabolism is relatively rapidly saturated, such that its kinetics proceeds from first to zero order; accordingly, the half-life of aspirin changes as a function of dose.
Acetaminophen is very commonly used in the pediatric population. Orally administered acetaminophen is rapidly (tmax = 1 hour) and nearly fully absorbed (>90%). In contrast, rectally administered acetaminophen is variable with mean bioavailability of 47% in children. Contrary to most NSAIDs, this drug has low protein binding. Its elimination occurs by conjugation to glucuronide and sulfates, the latter being the dominant form in children up to 9 years of age. A small amount of acetaminophen is oxidized by CYP enzymes. This latter pathway generates electrophilic compounds which require glutathione for detoxification; in overdose, this pathway is overutilized resulting in glutathione depletion and hepatic toxicity.
Specific COX-2 inhibitors have been approved by the Food and Drug Administration (United States) for a number of inflammatory and nociceptive indications; however, increased cardiovascular morbidity (and mortality) with a number of recently developed COX-2 inhibitors has resulted in limiting their applications particularly in adult patients (47,48). Selective COX-2 inhibitors, such as celecoxib,
rofecoxib, valdecoxib, as well as meloxicam (semi-selective COX-2 inhibitor), are readily absorbed enterally within 2 to 3 hours for the coxibs and 5 to 6 hours for meloxicam. Celecoxib, valdecoxib, and meloxicam are mainly metabolized by CYP2C9 and to some extent by CYP3A4 (49) and exhibit in the adult a t1/2 of 11 and 20 hours, respectively. In children, celecoxib is also well absorbed orally but is cleared much faster with a t1/2 of approximately 4 hours (50). Rofecoxib is metabolized by CYP1A2 and cytosolic liver enzymes (51) and has a t1/2 of 17 hours; its disposition in children is reported to be similar to that in adults (52).
rofecoxib, valdecoxib, as well as meloxicam (semi-selective COX-2 inhibitor), are readily absorbed enterally within 2 to 3 hours for the coxibs and 5 to 6 hours for meloxicam. Celecoxib, valdecoxib, and meloxicam are mainly metabolized by CYP2C9 and to some extent by CYP3A4 (49) and exhibit in the adult a t1/2 of 11 and 20 hours, respectively. In children, celecoxib is also well absorbed orally but is cleared much faster with a t1/2 of approximately 4 hours (50). Rofecoxib is metabolized by CYP1A2 and cytosolic liver enzymes (51) and has a t1/2 of 17 hours; its disposition in children is reported to be similar to that in adults (52).
Antipyretic Efficacy of NSAIDs and Acetaminophen
Fever represents the major indication of NSAIDs and acetaminophen in children. Of the NSAIDs, ibuprofen, approved in the United States for this indication, is by far the most commonly utilized in developed countries; aspirin, which has been mostly abandoned because of the risk of Reye syndrome, is still used in underdeveloped parts of the world. Doses of 10 mg per kg ibuprofen and 15 mg per kg acetaminophen have been found to exert similar antipyretic efficacy (53). Higher doses of ibuprofen (specifically 10 compared to 5 mg per kg) were more effective in lowering higher core temperatures (54).
NSAIDs in Juvenile Rheumatoid Arthritis and Other Inflammatory Conditions of Childhood
A number of randomized trials have evaluated the efficacy of NSAIDs in juvenile rheumatoid arthritis [for review, see reference no. (46)]. Overall diverse NSAIDs have been found to be equivalently effective, consistent with observations in the adult. Response is also dependent upon duration of therapy and increases with the latter to achieve approximately 80% efficacy after 8 weeks of treatment. Prolonged treatment with NSAIDs as needed for juvenile rheumatoid arthritis seems to augment the risk of gastroduodenal injury to a prevalence of 50% or more (19). On the other hand, significant renal complications are uncommon (55). Pseudoporphyria is an infrequent complication of NSAIDs, essentially limited to naproxen; the condition resolves upon drug discontinuation. Selective COX-2 inhibitors seem to be as effective as nonselective COX inhibitors, but exert fewer gastrointestinal complications (56,57). Although acetaminophen acts principally via COX-2 (14,15), its anti-inflammatory properties are limited as is its efficacy in severe rheumatoid arthritis (1,24,58).
Aspirin is commonly utilized in Kawasaki disease, but its efficacy remains controversial; however, aspirin combined with intravenous immunoglobulins is effective in this condition (59), but in acute Kawasaki it may cause thromboembolism (60). Similarly, the benefits of ibuprofen on lung function in cystic fibrosis have been confirmed in randomized trials (61,62).
Adverse Effects Secondary to Nsaids and Acetaminophen
Adverse effects of NSAIDs in children are similar in nature to those described in adults. Ibuprofen seems to exert a 7.2/100,000 risk of gastrointestinal bleeding (63). Mild reversible renal impairment is also observed in 8% to 10% of patients (64); dehydration doubles the risk. Other potential adverse effects of ibuprofen include aseptic meningitis and increased susceptibility of enhanced infections associated with group A β-hemolytic streptococci in children with varicella.
Table 48.2 Contraindications for Use of Nonsteroidal Anti-Inflammatory Drugs (NSAIDs) | |||||||||
|---|---|---|---|---|---|---|---|---|---|
|
Acetaminophen is relatively safe. High doses are associated with hepatotoxicity. Limitation of acetaminophen doses to less than 60 mg per kg per day virtually abolishes the risk of hepatotoxicity, unless used with other hepatotoxic agents (65).
Overall, NSAIDs have a good tolerance record. None-theless, in certain cases, they should be contraindicated (Table 48.2). In light of the cardiovascular effects recently associated with long-term COX-2 inhibitors use in adult, the dosing, tolerance, and safety of selective COX-2 inhibitors in children may need to be revisited.
Nsaids for Patent Ductus Arteriosus of the Newborn
Patent ductus arteriosus (PDA) is a common complication of the preterm infant. Its incidence is inversely related to gestational age. A patent DA is essential for fetal well-being because it allows 90% of the right ventricular output to bypass the high-resistance pulmonary vascular bed in utero. Prostaglandins play a major role in maintaining ductal patency during fetal life (66). Of the prostaglandins, PGE2 is the most important ductus arteriosus (DA) relaxant. Accordingly, inhibition in prostaglandin formation favors ductal constriction. Two COX inhibitors, indomethacin and, more recently, ibuprofen (2006), have been approved for the treatment of ductal patency in North America.
Indomethacin
Indomethacin is successful in closing 70% to 90% of patent DA in preterm infants (67). However, the response is variable due to physiological constraints of the more immature DA (68) and differing volume of distribution (69). Accordingly, optimal therapeutic dosing is difficult to establish; acceptable doses range from 0.1 to 0.25 mg per kg administered every 12 to 24 hours.
Numerous randomized trials have been performed to date to evaluate the efficacy of indomethacin in DA closure. A review of the prophylactic (first 24-hour postnatal),
presymptomatic (mostly within 72 hours of age), and symptomatic (usually 8 to 10 days of age) trials has recently been published (70). Interestingly, the rate of success in DA closure is irrespective of the age of onset within ages studied. Despite its efficacy in the majority of preterm infants, response to indomethacin seems to be diminished in children of lowest gestational age. In these cases, it has been observed that PGE2 production resurges within 5 days of indomethacin treatment (∼36 hour) (71,72). To circumvent this problem, prolonged treatments (5 to 7 days) with indomethacin were found to minimize ductal reopening (73,74,75). Evaluation of longer term morbidities revealed that overall the duration of oxygen therapy, hospital stay, the rate of bronchopulmonary dysplasia, and of death were unaffected by indomethacin treatment. Similarly, the rate of necrotizing enterocolitis and of retinopathy of prematurity was not significantly altered with the surprising exception of the collaborative trial (67).
presymptomatic (mostly within 72 hours of age), and symptomatic (usually 8 to 10 days of age) trials has recently been published (70). Interestingly, the rate of success in DA closure is irrespective of the age of onset within ages studied. Despite its efficacy in the majority of preterm infants, response to indomethacin seems to be diminished in children of lowest gestational age. In these cases, it has been observed that PGE2 production resurges within 5 days of indomethacin treatment (∼36 hour) (71,72). To circumvent this problem, prolonged treatments (5 to 7 days) with indomethacin were found to minimize ductal reopening (73,74,75). Evaluation of longer term morbidities revealed that overall the duration of oxygen therapy, hospital stay, the rate of bronchopulmonary dysplasia, and of death were unaffected by indomethacin treatment. Similarly, the rate of necrotizing enterocolitis and of retinopathy of prematurity was not significantly altered with the surprising exception of the collaborative trial (67).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


