Introduction to Non-Mendelian Inheritance
Non-Mendelian disorders have patterns of inheritance which do not conform to Mendel’s Law of Segregation where each ovum or sperm receives only one copy of a pair of genes. Important examples include mitochondrial inheritance, genetic imprinting, and multifactorial inheritance.
Mitochondrial disorders can be caused by abnormalities in either nuclear genes or in mitochondrial DNA. Mitochondrial disorders caused by abnormalities of nuclear genes are inherited in a Mendelian fashion. Mutations in mitochondrial DNA (mtDNA) cause disorders which have mitochondrial inheritance. An important feature of disorders with mitochondrial inheritance is heteroplasmy, i.e., varying amounts of mutant and normal mtDNA in each cell. Mitochondrial DNA is almost exclusively maternally transmitted; hence, mitochondrial disorders do not follow Mendel’s Law of Segregation as all children of an affected mother will inherit abnormal mtDNA. The manifestations of disorders with mitochondrial inheritance may vary widely among affected offspring because different amounts of normal and mutant mtDNA are present in each ovum.
Genetic imprinting is responsible for another class of non-Mendelian disorders. Several imprinted genes are normally expressed or silenced depending on whether they have been paternally or maternally transmitted. Chemical modifications to the gene structure but not the gene sequence lead to this phenomenon. Risk of recurrence of an imprinted disorder depends on the specific genetic mechanism leading to aberrant gene imprinting and the parent of origin.
Disorders with multifactorial inheritance are caused by the effects and complex interactions of multiple susceptibility genes, each usually with a relatively small effect, and environmental and epigenetic factors. Multifactorial conditions constitute a significant fraction of common birth defects such as congenital heart disease, neural tube defects, facial clefting, and common medical problems such as diabetes, autoimmune disease, cancer, and most cases of autistic spectrum disorder. This class of non-Mendelian disorders is the most etiologically complex and poorly understood. While multiple members of the same family are sometimes affected, multifactorial disorders are not associated with Mendelian patterns of inheritance.
Features of Mitochondrial Inheritance
1. Mitochondrial DNA (mtDNA) is almost exclusively maternal in origin; there is a negligible contribution from sperm.
2. There is exclusive maternal transmission.
3. Only maternal offspring are affected.
4. Males and females are equally likely to be affected.
5. No transmission occurs to descendants of affected males.
6. Phenotypic predictions are complicated by heteroplasmy (mixture of normal and abnormal mtDNA).
Mitochondrial Disorders
A couple is referred for genetic counseling because their 14-month-old daughter has just been diagnosed with Leigh syndrome. Over the past several months, she had regression of motor skills with onset of abnormal movements, progressive hypotonia, irritability, and loss of appetite with vomiting. Seizures began at 10 months of age and symptoms seemed to worsen in association with viral illnesses. She has elevated lactate concentrations in blood and cerebrospinal fluid. Brain imaging showed multiple spongiform lesions in specific areas of the brain, which are findings characteristic of Leigh syndrome. The wife, age 33 years, is now 10 weeks pregnant.
Leigh syndrome, one of a number of mitochondrial disorders associated with defective energy metabolism, is a neurodegenerative disorder which results from a deficiency of any of the mitochondrial respiratory chain complexes. Mutations in a number of different genes with different inheritance patterns can lead to the Leigh syndrome phenotype.
About 70% of cases of Leigh syndrome are associated with mutations in nuclear DNA (nDNA) and have autosomal or X-linked inheritance patterns (i.e., they are inherited as autosomal recessive, autosomal dominant, or X-linked conditions). The remaining 30% of cases are attributed to mutations in mitochondrial DNA (mtDNA) and have an inheritance pattern which is characteristic of mitochondrial inheritance.
Mitochondrial inheritance differs from Mendelian inheritance because functioning mitochondria and the mtDNA within them are inherited exclusively from one’s mother. If a woman has a mitochondrial disorder due to a mtDNA mutation, each of her children, regardless of sex, has close to a 100% chance of inheriting the disorder if the mutant mtDNA are present in her ova. However, the severity of disease expression in her children may be highly variable due to differing amounts of mutant mtDNA load. Normal mitochondrial functioning and a normal phenotype may be present if the mutant mtDNA load is below a certain threshold. Males with mitochondrial disorders due to mutant mtDNA do not transmit the disorder to their children. Evidence for transmission of disease from males to their offspring essentially excludes mitochondrial inheritance from consideration.
A mitochondrial disorder caused by a mtDNA mutation may arise de novo in the mitochondria of an affected child. Almost always, however, mitochondrial disorders due to mtDNA mutations are transmitted from the mother who also has the mtDNA mutation. She will usually be asymptomatic or less severely affected.
There is a wide continuum of disease severity within and among families affected by mitochondrial disorders due to mtDNA mutations. This is largely accounted for by heteroplasmy, the situation in which there is more than one type of mtDNA present within each cell. Hundreds of copies of mitochondria and mitochondrial DNA are present in each human cell.
The burden of mutant mtDNA varies between cells and among different tissues. A high burden of mutant mtDNA will usually result in early onset and severe disease. A low burden of mutant mtDNA may be compatible with a normal phenotype, with milder symptoms, or with onset of symptoms later in life. In addition to heteroplasmy, the nature of the mutation itself will also influence the severity of disease expression of mitochondrial disorders.
Once the disorder is suspected from the clinical presentation, further diagnostic testing may include brain imaging, metabolic studies of blood and cerebrospinal fluid, histopathology of muscle biopsy, and respiratory chain enzyme studies of tissue biopsies.
Establishing the molecular basis of a mitochondrial disorder will provide information about recurrence risk in future children, allow for prenatal diagnosis in future pregnancies, and permit the identification of at-risk relatives. However, molecular testing is complicated by the extensive genetic heterogeneity of Leigh syndrome; at least six autosomal genes and 11 mitochondrial genes are associated with the syndrome. Furthermore, for some mtDNA mutations that cause Leigh syndrome, the percentage of abnormal mtDNA may vary between different tissues and may decrease in white blood cells with increasing age. Analysis of mtDNA obtained from skeletal muscle or urine sediment is associated with the highest sensitivity in the detection of mtDNA mutations for Leigh syndrome. For analysis of nDNA mutations when a nuclear mitochondrial disorder is suspected, DNA obtained from leukocytes is satisfactory.
Obtaining information about the family may provide clues as to the mode of inheritance and can help direct the strategy for molecular testing. For disorders due to mtDNA mutations, affected relatives on the maternal side may have symptoms that have not been recognized as part of the disease spectrum. In the absence of a family history that suggests a mitochondrial disorder, an autosomal recessive disorder or a de novo autosomal dominant disorder would be possibilities.
A family history is collected. Neither parent reports relatives with neurodegenerative disorders. The father of the child with Leigh syndrome is in good health. The mother’s health history is remarkable only for migraine headaches. Her brother developed night blindness in his late teens and is thought to have retinitis pigmentosa. He has four children who are healthy. The maternal grandmother died at age 62 years shortly after being diagnosed with a cardiac arrhythmia.
The mother’s health and family history suggest that a mtDNA mutation could be the underlying cause for her daughter’s Leigh syndrome. Migraine headaches, night blindness, diabetes, cardiac conduction defects leading to sudden unexpected death, and muscle weakness are included in the spectrum of clinical problems that can be seen in the mothers and other maternal relatives of a child with Leigh syndrome.
Mutations in one of the mtDNA genes causing Leigh syndrome can also lead to another disorder known as NARP (neurogenic muscle weakness, ataxia, and retinitis pigmentosa). Other features of NARP which may be present include short stature, sensorineural hearing loss, progressive external ophthalmoplegia, and cardiac conduction defects. The symptoms of NARP often begin in early childhood, may remain stable for many years, may be exacerbated by viral illnesses, or may not be appreciated until adulthood. In some families, affected individuals may have classic Leigh syndrome, NARP, or any combination of the features of these disorders.
Analysis of mtDNA obtained from the affected daughter’s skeletal muscle revealed a mtDNA mutation associated with both Leigh syndrome and NARP. This mutation was subsequently documented in the mother and her brother. The couple wants prenatal diagnosis of Leigh syndrome in the current pregnancy.
Each of the couple’s children will inherit both mutant and wildtype mtDNA from the mother that will be present in varying proportions. A number of different options are available to decrease transmission of mtDNA mutations (see Table 3.1). The ratios of wildtype and mutant DNA present in uncultured chorionic villi or amniocytes reflect the mutant load present in most tissues at birth. Attempts have been made to predict the severity of disease in a fetus based on the proportions of mutant and wildtype mtDNA. These predictions should be made cautiously but appear, at least for Leigh syndrome, to provide reasonable guidance if there are very high or very low proportions of mtDNA in the fetal sample. Prior to initiating prenatal diagnosis, couples should be aware that predictions about the phenotypic outcome would not be possible when the proportions of mutant and wildtype mtDNA are roughly equivalent. Analysis of mtDNA for prenatal diagnosis should be performed on uncultured chorionic villi or amniocytes due to the possibility that the proportions of wildtype and mutant mtDNA will change during cell culture.
Table 3.1 Strengths and weaknesses of reproductive options for preventing transmission of mtDNA mutations
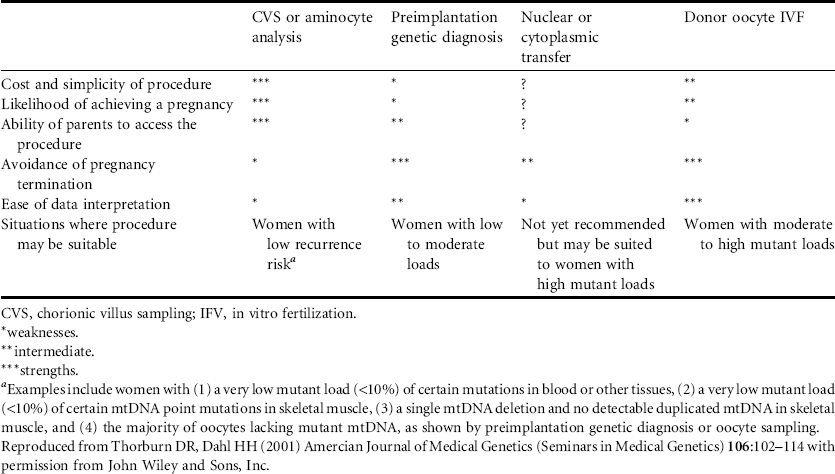
In vitro fertilization with preimplantation genetic diagnosis would also be possible for future pregnancies. Only embryos which have a very low or preferably zero mutant load should be used for implantation. In vitro fertilization using an ovum from a donor who is not a maternal relative of the wife would be another alternative to avoid the risk of having a child affected by Leigh syndrome. Research has also showed the feasibility of using the nucleus of a woman’s ovum and cytoplasm with mitochondria from another woman’s ovum for the maternal contribution to a conceptus.
Further Reading
1. Bredenoord AL, Pennings G, Smeets HJ et al. (2008) Dealing with uncertainties: ethics of prenatal diagnosis and preimplantation genetic diagnosis to prevent mitochondrial disorders. Human Reproduction Update 14:83–94.
2. Thorburn DR, Dahl, HH. (2001) Mitochondrial disorders: genetics, counseling, prenatal diagnosis and reproductive options. American Journal of Medical Genetics (Seminars in Medical Genetics) 106:102–114.
The parents of a 4-year-old girl who may have Angelman syndrome are seen for genetic counseling. The daughter has the characteristic findings of the disorder which include developmental delay that became apparent in the first year of life, severe mental retardation, absent speech, a distinctive facial appearance, hand flapping, and an unusually happy demeanor. She also has microcephaly and seizures that are commonly seen in Angelman syndrome. The mother is pregnant and wants to know the risk of recurrence of her daughter’s problems and whether prenatal diagnosis is available. The couple has two other children who are developmentally normal. The daughter’s diagnosis has not been confirmed by molecular testing. The mother is 8 weeks pregnant.
Angelman syndrome is a disorder resulting from abnormal genetic imprinting of the UBE3A gene on chromosome 15q11.2-q13. Imprinting occurs when the parental origin of a gene determines whether or not it is expressed. Only a small minority of human genes are subject to imprinting effects. Imprinting results from epigenetic phenomena in which gene expression is controlled by DNA methylation and other chemical processes that alter the DNA but not the gene sequence. In general, genes that are methylated are silenced and demethylation results in activation of gene expression. In each generation, reprogramming (methylation or demethylation) of imprinted genes occurs in ova and sperm. The sex of the transmitting parent determines whether or not an imprinted gene will be expressed or silenced in the next generation, regardless of whether that gene was expressed in that parent.
In normal individuals, only the maternally inherited UBE3A gene is expressed while the paternally inherited allele is silenced. Angelman syndrome arises due to loss of expression of the maternally inherited UBE3A allele, which can be a consequence of different genetic processes that include the following.
1. A chromosomal deletion at chromosome 15q11.2-q13 on the maternally inherited allele accounts for about 70% of cases. Most are de novo. Almost all are microdeletions that cannot be detected by routine metaphase karyotyping but can be found by fluorescence in situ hybridization (FISH) or array comparative genomic hybridization (array CGH). In rare cases, microdeletions are found in association with either de novo or inherited chromosomal rearrangements involving the 15q11.2-q13 locus.
2. Seven percent of cases are due to paternal uniparental disomy for chromosome 15, where both copies of chromosome 15 are from the father and there is absence of the maternally inherited chromosome 15. Uniparental disomy can be detected by analysis of DNA polymorphisms on chromosome 15 in the affected child and parents and by other molecular techniques.
3. Three percent of cases are due to a 6–200 kb microdeletion in the imprinting center of the UBE3A gene on the maternally inherited chromosome which results in silencing of the gene. This explanation is inferred if aberrant gene methylation is present and a chromosomal deletion and uniparental disomy have been excluded.
4. Another 11% of cases have a point mutation in or deletion of the entire UBE3A gene which can be detected by gene sequencing or other molecular methods used to detect chromosomal deletions.
5. At this time, an underlying genetic abnormality is not found in about 11% of individuals with the clinical diagnosis of Angelman syndrome. This may be due to an incorrect clinical diagnosis, or other unidentified genetic mechanisms resulting in aberrant UBE3A gene function or expression.
Chromosomal microdeletions, uniparental disomy, and UBE3A gene imprinting center defects are all associated with abnormal DNA methylation. However, abnormal DNA methylation does not establish the underlying molecular basis of the disorder, information which is important for providing information about the risk of recurrence and accurate prenatal diagnosis. If abnormal DNA methylation is present, array CGH or FISH should be performed to look for a chromosomal deletion. If a chromosome deletion is found, a metaphase karyotype should be obtained to look for a chromosome rearrangement. If FISH or array CGH is normal, uniparental disomy studies should be performed. If uniparental disomy is not found, further molecular studies could be performed to identify a small microdeletion or other change in the imprinting center of the UBE3A gene.
If DNA methylation studies are normal, UBE3A gene sequence analysis is indicated.
Risk of recurrence of Angelman syndrome varies widely depending on the underlying molecular pathogenesis. As shown in Table 3.2, risk of recurrence is small unless a parent carries a chromosomal rearrangement involving chromosome 15, or if the mother herself carries a UBE3A gene mutation or imprinting center defect. (For these latter possibilities, the mother herself could be unaffected by Angelman syndrome because the UBE3A gene mutation could either have been transmitted from her father or have arisen de novo on her paternally inherited chromosome 15.) For de novo mutations in the child, the risk of recurrence is very small, but not zero because of the possibility of gonadal mosaicism.
Array CGH reveals a microdeletion at the chromosome 15q12 locus, confirming the clinical diagnosis of Angelman syndrome. Routine cytogenetic analysis of the daughter reveals an apparently balanced translocation between chromosomes 4p16 and 15q12.
Table 3.2 Genetic mechanisms in Angelman syndrome (AS) and Prader–Willi syndrome (PWS) and risk of recurrence in the siblings of an affected child
| Genetic mechanism | Percentage of families | Recurrence risk |
| Deletion of 15q11-q13 | 70 (both AS and PW) | <1% |
| UPD of chromosome 15 | ∼25 (PWS) 3–7 (AS) | <1% |
| Mutation in UBE3A | ∼10 (AS) | 50% if present in mother |
| No identifiable molecular abnormality | ∼10 (AS) | Unknown |
| Imprinting center defect without mutation in the imprinting center | 1–3 (both) | <1% |
| Imprinting center defect with mutation in the imprinting center | <1 (both) | 50% if present in father (PWS) or mother (AS) |
| De novo unbalanced rearrangement of 15q11-q13 | <1 (both) | <1% |
| Inherited unbalanced rearrangement of 15q11-q13 | <1 (both) | ≤50% |
| UPD due to parental rearrangement involving chromosome 15 | <1 (both) | Depends on translocation; if parent has 15;15 Robertsonian translocation, risk would approach 100%; for most others, risk is ≤0.75% |
| UPD, uniparental disomy. Data from Williams CA, Dagli AI, Driscoll DJ (Updated 9/5/2008). Angelman syndrome. In: Gene Reviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009; Cassidy SB, Schwartz S (Updated 3/24/2008). Prader-Willi syndrome. In: Gene Reviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009. Table adapted from Kokkonen H. (2003) Genetic changes of chromosome region 15q11–q13 in Prader-Willi and Angelman syndromes in Finland http://herkules.oulu.fi/isbn9514270274/html/index.html | ||
The translocation could have arisen as a de novo event or have been inherited from one of the parents who are both phenotypically normal.
The father has a normal karyotype. The mother has a balanced translocation between chromosomes 4p16 and 15q12. Array CGH does not show any evidence for a deletion or duplication of genetic material.
The daughter inherited her mother’s chromosomal translocation which, in contrast to the mother, also has a small interstitial deletion on chromosome 15 resulting in absence of a functional maternally derived UBE3A gene. The identification of the chromosomal translocation in the mother has implications both for recurrence of conceptuses who have unbalanced translocations involving partial trisomies and monosomies for chromosomes 4 and 15 and Angelman syndrome due to an interstitial deletion at the 15q12 locus.
This family is an example of the rare circumstance in which the same apparently balanced translocation is associated with a normal phenotype in one relative and an abnormal phenotype in another due to a de novo structural alteration, in this case an interstitial deletion at one of the translocation breakpoints. In the current pregnancy, the risk of recurrence of another child who has inherited the mother’s apparently balanced translocation and also has a microdeletion at the chromosome 15q12 locus is very small. This risk is far outweighed by the significant chance of a child with an unbalanced chromosomal translocation.
For individuals who carry either a reciprocal or Robertsonian translocation involving chromosome 15, there is also a small additional risk of uniparental disomy for chromosome 15 resulting from malsegregation of the chromosomes involved in the translocation during meiosis. This could lead to some gametes with two copies of chromosome 15 and some gametes with no copy of chromosome 15. Uniparental disomy for chromosome 15 can arise via two mechanisms. The more likely situation would be trisomy rescue in which an embryo with trisomy 15 is “rescued” by loss of a chromosome 15 after fertilization, resulting in a karyotypically normal fetus who now has two copies of either the maternally or paternally derived chromosome 15. Another, less likely, situation would be a zygote conceived with one gamete with disomy for chromosome 15 and another which was nullisomic for chromosome 15. Both of these situations could result in absence of either the maternally or paternally inherited chromosome 15, leading to Angelman syndrome or Prader–Willi syndrome, respectively. The risk of uniparental disomy in the setting of a parent with a balanced translocation is estimated at 0.7%.
If other maternal relatives carry the translocation, they would face similar risks.
The woman’s brother, who is healthy and developmentally normal, is referred for consultation because he also carries the chromosome translocation between chromosomes 4p16 and 15q12. Array CGH does not detect a duplication or deletion of genetic material.
This man is at significant risk for conceiving children who have unbalanced chromosomal complements involving various combinations of partial monosomies and partial trisomies for chromosomes 4 and 15. In addition, there would be a very small additional risk of a child with a de novo submicroscopic interstitial deletion at the site of one of the translocation breakpoints as discovered in his niece. Such a deletion on chromosome 15q12 would not result in Angelman syndrome because it would be of paternal origin and the affected child would still have a functional maternally inherited UBE3A gene. However, the 15q11.2q13 locus also contains the Prader–Willi critical region (PWCR) which contains genes which are subject to imprinting effects. Genetic abnormalities in the paternally derived PWCR will result in Prader–Willi syndrome. The main features of Prader–Willi syndrome include neonatal and infantile hypotonia and feeding problems, mild to moderate mental retardation, hyperphagia during childhood leading to obesity, short stature, small hands and feet, hypogonadism, and distinctive facial features.
Almost all individuals with Prader–Willi syndrome have an imprinting defect in the PWCR which is caused by one of several mechanisms leading to loss of expression of genes in the paternally derived PWCR. About 75% of cases have chromosomal microdeletions on the paternally derived allele (associated with abnormal DNA methylation and also diagnosable by FISH or array CGH). Maternal disomy for chromosome 15 (associated with abnormal DNA methylation and diagnosable by molecular analysis) is present in about 25% of cases. Less than 1% of cases are due to an imprinting center defect in the paternally derived chromosome. Imprinting center defects can be caused by microdeletions in the paternal PWCR imprinting center, which are associated with abnormal DNA methylation and can be detected by gene sequencing, or, more commonly, they can be due to abnormalities of gene methylation of the paternal allele which are not associated with a gene sequence change. While abnormal methylation confirms the clinical diagnosis of Prader–Willi syndrome, establishing the underlying molecular basis for the disorder is important for recurrence risk counseling and accurate prenatal diagnosis.
As shown in Table 3.2, inherited chromosomal rearrangements are a rare cause of Angelman syndrome and Prader–Willi syndrome and the presence of both disorders in the same family is an extremely unusual situation.
The mother who carries the balanced reciprocal translocation between chromosomes 4p16 and 15q12 elects to undergo chorionic villus sampling based on the increased risk of conceptuses with unbalanced chromosomal complements and the chance of recurrence of the microdeletion at the chromosome 15q12 locus. The karyotype of cultured chorionic villus cells reveals that the fetus inherited the mother’s apparently balanced translocation between chromosomes 4p16 and 15q12.
As noted above, most microdeletions causing Angelman syndrome, including the one found in the woman’s affected daughter, cannot be detected by routine metaphase karyotyping. Two approaches, FISH or array CGH, can be used to investigate the possibility of a submicroscopic microdeletion at the Angelman syndrome locus on chromosome 15q12. FISH uses probes specific for this locus and would address the presence of absence of a deletion (or duplication) at the Angelman syndrome locus only. However, this woman’s reciprocal translocation also involves a breakpoint on chromosome 4p16. Inherited balanced rearrangements are rarely associated with submicroscopic de novo duplications or deletions resulting in an abnormal phenotype, although this did occur at the chromosome 15q12 locus for the daughter. Array CGH of DNA obtained from cultured or uncultured chorionic villi would also address this risk of a duplication or deletion of genetic material at the chromosome 4p16 locus, as well as the 15q12 locus. Because the early embryo and placenta are hypomethylated, it is not yet certain whether DNA methylation studies would be reliable in the first trimester.
Within the limits of the technology, array CGH did not detect a deletion or duplication at the breakpoints of the translocation on chromosomes 4 and 15 or elsewhere in the fetal genome.
Because the maternal translocation involving chromosome 15 has a small chance of resulting in maternal uniparental disomy for chromosome 15, which causes Prader–Willi syndrome, molecular studies are indicated to determine whether both parents have contributed a chromosome 15.
In future pregnancies, preimplantation genetic diagnosis using probes for the translocation and the typical Angelman syndrome deletion could be used. Whether uniparental disomy for chromosome 15 could be addressed at the same time would depend on available technology and experience in the laboratory. Because the early embryo is hypomethylated, preimplantation genetic diagnosis by methylation studies is not recommended.
Similar approaches could be used for the woman’s brother and his wife for a future pregnancy to address the risks of a fetus with an unbalanced chromosomal translocation, the possibility of a microdeletion at chromosome 15q12 leading to Prader–Willi syndrome, and paternal uniparental disomy leading to Angelman syndrome.
Further Reading
1. Cassidy SB, Schwartz S (Updated 3/24/2008) Prader-Willi syndrome. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009.
2. Gurrieri F, Accadia M (2009) Genetic imprinting: the paradigm of Prader-Willi and Angelman syndromes. Endocrine Development 14:20–28.
3. Horsthemke B, Maat-Kievit A, Sleegers E et al. (1996) Familial translocations involving 15q11-q13 can give rise to interstitial deletions causing Prader-Willi or Angelman syndrome. Journal of Medical Genetics 33:848–851.
4. Knoll JH, Wagstaff J, Lalande M (1993) Cytogenetic and molecular studies in the Prader-Willi and Angelman syndromes: an overview. American Journal of Medical Genetics 46 (1):2–6.
5. Smeets, DFCM, Hamel BCJ, Nelen, MR et al. (1992) Prader-Willi syndrome and Angelman syndrome in cousins from a family with translocation between chromosomes 6 and 15. New England Journal of Medicine 326 (12):807–811.
6. Stalker HJ, Williams CA (1998) Genetic counseling in Angelman syndrome: the challenges of multiple causes. American Journal of Medical Genetics 77: 54–59.
7. Williams CA, Dagli AI, Driscoll DJ (Updated 9/5/2008) Angelman syndrome. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009.



