Introduction to Mendelian Inheritance
Disorders with Mendelian inheritance, which are also known as single gene disorders, are due to major effects of one or both members of a pair of genes. The separation of these genes during meiosis with each ovum or sperm receiving only one copy of that gene is known as Mendel’s Law of Segregation. Thus, the genes which cause Mendelian disorders are transmitted in a predictable pattern from one generation to the next. There are rare circumstances in which single gene disorders can arise in violation of Mendelian inheritance. Uniparental disomy in which both copies of a gene are transmitted from one parent is one example. However, Mendelian inheritance still applies for the vast majority of families in which a single gene disorder is present.
Hundreds of Mendelian disorders are amenable to prenatal diagnosis by molecular analysis. Counseling about the results of prenatal diagnostic testing for these conditions is usually straightforward when the disorder is consistently associated with a narrow range of clinical abnormalities, the diagnosis of the affected family members has been clearly established, or the nucleotide change(s) that have been identified in a gene are already known to be pathogenic. Such an example is Tay–Sachs disease, where homozygosity or compound heterozygosity for common mutations in the HEXA gene invariably results in a fatal neurodegenerative course during the first years of life.
However, such ideal counseling situations are often not the reality. For some disorders, such as X-linked adrenoleukodystrophy, a devastating neurodegenerative course during the first decade of life may affect one family member while, in another, milder neurologic impairment develops only in adulthood. Such a wide spectrum in severity and age of onset of symptoms among family members who have the same mutation are also the hallmarks of many autosomal dominant disorders, as illustrated by tuberous sclerosis. Phenotypic heterogeneity can also be seen in autosomal recessive disorders such as spinal muscular atrophy in which known and unknown genetic modifiers influence severity of disease. Although prenatal diagnosis of a disorder is highly accurate once a disease causing mutation(s) has been identified in a family, precise predictions about expression of the phenotype are sometimes not possible to make and complicate decision making about whether to pursue prenatal diagnosis or consider pregnancy termination.
For some disorders, counseling is complicated by the identification of a novel nucleotide change of uncertain pathologic significance in an affected family member. Establishing the pathogenicity of such an unreported nucleotide change may not be possible, thus leaving doubt about the accuracy of preimplantation or prenatal genetic diagnosis.
Another obstacle to providing accurate risk estimates is the phenomenon of genetic heterogeneity where mutations in more than one gene can lead to the same or similar phenotype. At times, there may even be a different mode of inheritance. Incorrect assumptions about an inheritance pattern can lead to erroneous assessments of risk in a fetus and missed opportunities for prenatal diagnosis. If confirmatory clinical and/or diagnostic laboratory testing has not been accomplished for an affected relative, the estimation of risk to a fetus must be made cautiously.
These challenges and basic concepts related to single gene inheritance including pedigree interpretation, calculation of risk estimates, the Hardy–Weinberg equilibrium, atypical X-linked inheritance, factors influencing expression of an X-linked disease in females, and trinucleotide repeat expansions are presented here.
Features of Autosomal Dominant Inheritance
1. The trait (phenotype) appears if the abnormal gene is present on only one homologous autosomal chromosome.
2. Each child of an affected individual has a 50% chance of inheriting the gene.
3. Males and females are affected with equal frequency and transmit the abnormal gene to sons and daughters with equal frequency.
4. Family members who do not carry the abnormal gene do not transmit the trait to their children.
5. There can be a wide range in the severity and age of onset of the trait (variable expression).
6. For some autosomal dominant disorders, a percentage of individuals with the abnormal gene do not express the trait although they can transmit it, which then gives the appearance of “skipped” generations (incomplete penetrance).
Autosomal Dominant Polycystic Kidney Disease
A couple comes for genetic counseling because the 31-year-old husband reports that he has polycystic kidney disease diagnosed by renal imaging performed as a teenager after his father developed end-stage renal disease at age 40 years due to polycystic kidney disease. His father and paternal grandfather died of a stroke in their early 40s. It is not known whether the paternal grandfather had polycystic kidney disease. The husband is an only child, as was his father. The husband’s mother is alive at age 78 years. The husband reports that he has hypertension, which is well controlled by antihypertensive medications, and that he has regular brain imaging by MRI. The couple wants to avoid the birth of a child who will develop polycystic kidney disease.
Autosomal dominant polycystic kidney disease (ADPKD) is one of the most common single gene disorders, affecting about 1 in 800 individuals. The disorder, which usually begins in mid-life, often has extrarenal manifestations which include cysts in the liver, pancreas, and seminal vesicles. In addition, there is an increased risk of intracranial aneurysm, stroke and dissection of the thoracic aorta due to weakening of the arterial wall.
Mutations in two genes, PKD1 and PKD2, account for 85% and 15% of cases, respectively. Mutations in other gene(s) may cause disease in rare families in whom no identifiable mutation in PKD1 or PKD2 can be detected and no linkage to either of these genes can be established.
The variable expression and pleiotropic effects of the disorder can in part be explained by the underlying molecular pathogenesis, as well as other, yet unidentified, genetic and environmental factors. Compared with PKD2 gene mutations, PKD1 gene mutations are associated with more severe disease expression including earlier age of onset, an increased chance of associated complications, earlier onset of end-stage renal failure, and earlier age at death. For example, almost all carriers of a PKD1 gene mutation, but only two-thirds of PKD2 gene mutation carriers, will have renal cysts evident on ultrasonographic examination by age 30 years.
Gene sequencing and duplication-deletion testing of PKD1 and PKD2 are available through clinical diagnostic laboratories.
The husband submits a blood sample for molecular testing of his PKD1 and PKD2 genes. No pathogenic nucleotide changes were found in his PKD2 genes. Analysis of his PKD1 genes identified a nucleotide change involving a transversion from guanine to cytosine at codon 2266. The laboratory report states that this finding could be either a benign polymorphism or a disease-associated mutation, but did not provide further information.
A number of criteria can be used to help distinguish a pathogenic mutation from a silent, benign polymorphism. These include the following:
1. The nucleotide change has been reported in other families with ADPKD by searching the Polycystic Kidney Disease Mutation Database which is accessible by internet and updated frequently.
2. The nucleotide change segregates with PKD in the family. Do all affected family members have the nucleotide change while unaffected family members do not?
3. The nucleotide change is predicted to cause a significant alteration in the protein structure. Major changes in the structure of the protein are often associated with abnormalities of function.
4. The normal DNA sequence has remained the same among different mammalian and other species, suggesting, in evolutionary conservation terms, that it is very important for normal functioning.
Review of the Polycystic Kidney Disease Mutation Database showed that the nucleotide change in the husband has not been reported in other affected individuals. Demonstrating segregation of the nucleotide change with ADPKD in the husband’s family is not possible as there are no other living affected family members.
However, two lines of evidence give fairly strong support that the nucleotide change found in the husband is a disease-associated mutation. First, the DNA alteration is predicted to substantially alter the protein product of the gene based on modeling programs by biochemists. Second, the normal DNA sequence at codon 2266 in the PKD1 gene is highly conserved among different mammalian species and among fish.
Whether the couple should proceed with preimplantation or prenatal genetic diagnosis for ADPKD based on the available information suggesting that the nucleotide change is pathogenic requires careful consideration. The evidence for pathogenicity is good but not definitive. Before proceeding with a pregnancy, testing of the husband’s unaffected mother to determine whether she has the nucleotide change or normal gene sequence would provide some additional information. If she has the nucleotide change present in her son, this would be strong evidence that it is not disease-causing. Absence of the nucleotide change in the mother would show that it was transmitted from his affected father, barring new mutation or non-paternity, and provides evidence of segregation of the nucleotide change with disease status. To prove in a statistically meaningful way that a nucleotide variant segregates with disease status, many affected and unaffected individuals in a family would need to be studied, something that is not possible given the very small size of the husband’s family.
The husband’s mother has DNA testing which shows that she does not carry the PKD1 gene nucleotide change identified in her son.
Information about the mother provides further support for the pathogenicity of the nucleotide change. Unfortunately, no further testing is currently available that can provide definitive information for this family. As more information is gathered into DNA databases about ADPKD families, it is possible that other families will be reported with a similar gene alteration. This information may not be forthcoming for several years, and the couple will need to make reproductive decisions with less than perfect information.
Further Reading
1. Brun M, Maugey-Laulom B, Eurin D et al. (2004) Prenatal sonographic patterns in autosomal dominant polycystic kidney disease: a multicenter study. Ultrasound in Obstetrics and Gynecology 24:55–61.
2. Harris PC, Torres VE (Updated 6/2/2009) Polycystic Kidney Disease, Autosomal Dominant. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009.
3. Pei Y (2003) Molecular genetics of autosomal dominant polycystic kidney disease. Clinical and Investigative Medicine 26 (5):252–258.
4. Vora N, Perrone R, Bianchi DW (2008) Reproductive issues for adults with autosomal dominant polycystic kidney disease. American Journal of Kidney Disease 51 (2):307–318.
5. Wilson PD (2004) Polycystic kidney disease. New England Journal of Medicine 350:151–164.
Hereditary Colon Cancer
A 25-year-old man is newly married and comes for genetic counseling with his wife to learn about reproductive options. The man has known for several years that he carries a mutation in one of his MLH1 genes, one of the genes associated with hereditary non-polyposis colon cancer (HNPCC). He had testing as a teenager at the urging of his mother who was diagnosed with colon cancer at age 30 years. The man’s family history also includes a maternal aunt, maternal grandmother and maternal great grandfather who were all diagnosed with colon cancer in their 30s. The man has yearly colonoscopies.
Mutations in the MLH1 gene, one of the genes involved in DNA mismatch repair, are associated with an increased risk of HNPCC. Individuals who carry a DNA mismatch repair mutation have a lifetime risk of developing HNPCC of about 80% as well as increased risk of other cancers including cancers of the endometrium, ovary, stomach, small intestine, hepatobiliary tract, upper urinary tract, brain, and skin. HNPCC has autosomal dominant inheritance. Five percent of all cases of colon cancer are due to HNPCC.
Each of the couple’s children has a 50% risk of inheriting his MLH1 gene mutation and a high lifetime risk of cancer. If the couple wants to avoid the birth of a child with the MLH1 gene mutation, preimplantation genetic diagnosis could be utilized to select for embryos which are unaffected. Prenatal diagnosis by chorionic villus sampling or amniocentesis could also be performed although most couples are not inclined to consider pregnancy termination for conditions that usually do not have manifestations until mid-life. Another alternative would be artificial insemination with sperm from an unrelated donor or an unaffected male relative of the husband. If the sperm donor is related to the husband, he should be screened for the familial MLH1 gene mutation. An unrelated sperm donor could also be screened for an MLH1 gene mutation although the chance of such a donor having that mutation would be extremely low, and the MLH1 gene is only one of many genes whose mutations carry a high lifetime cancer risk.
The couple states that they are being strongly encouraged to have preimplantation genetic diagnosis by the husband’s family to eliminate the risk of HNPCC in future generations. They, however, state that they are much more inclined to conceive a child naturally and hope that better therapies for cancer prevention are developed by the time their children reach adulthood.
Further Reading
1. Kastrinos F, Stoffel EM, Balmaña J et al. (2007) Attitudes toward prenatal genetic testing in patients with familial adenomatous polyposis. American Journal of Gastroenterology 102 (6):1284–1290.
2. Spits C, DeRycke M, Van Ranst N et al. (2007) Preim-plantation genetic diagnosis for cancer pre-disposition syndromes. Prenatal Diagnosis 27 (5):447–456.
3. Tops CM, Wijnen JT, Hes FJ (2009) Introduction to molecular and clinical genetics of colorectal cancer syndromes. Best Practices and Research, Clinical Gastroenterology 23 (2):127–146.
Huntington Disease
A woman is referred for genetic counseling in the first trimester because the family history collected by her obstetrician includes relatives of her husband who reportedly have Huntington disease. His mother, maternal grandmother, two maternal aunts, and a female maternal first cousin have the disorder or have died from its complications. The husband states that his mother died at age 58 years after a 20-year course of the disease. Two maternal uncles are in their 60s and unaffected. The husband is 38 years old and in good health. He has two brothers who are reported to be well in their 30s.
Huntington disease is an autosomal dominant neurodegenerative disorder usually characterized by progressively worsening involuntary movements, cognitive impair-ment, and psychiatric disturbance. There is a wide range in age of onset from early childhood to old age, although the majority of affected individuals develop symptoms between ages 30 and 50 years. The molecular basis of Huntington disease is a trinucleotide repeat expansion in one copy of the huntingtin gene in which the number of CAG repeats that exceed a certain threshold will eventually result in manifestations of the disorder. Age of onset of symptoms is correlated with the size of the CAG expansion. Alleles with CAG expansions are unstable during male meiosis, resulting in the phenomenon of anticipation in which the offspring of affected males often have an earlier onset of symptoms due to large trinucleotide expansions in one of their Huntington disease genes.
Because other neurodegenerative disorders can occasionally mimic the symptoms of Huntington disease, it is important to confirm that at least one affected member of the family has had the clinical diagnosis confirmed by molecular testing to provide the most accurate genetic counseling, especially if the husband would be considering molecular testing of his own Huntington disease genes. If Huntington disease is the correct diagnosis for the husband’s relatives, he has a 50% chance of having inherited his mother’s abnormal Huntington disease gene and developing symptoms of the disorder at some point in his life.
A review of the affected maternal cousin’s molecular testing shows that she has one normal-sized Huntington disease gene and another allele with 42 CAG repeats (normal range < 36 CAG repeats). This result confirms the clinical diagnosis of Huntington disease in the husband’s family.
The couple is surprised to learn that the husband is at risk for developing Huntington disease. Because no males in his family are known to have the disorder, he and his relatives were under the strong impression that males were not susceptible.
The distortion in the sex distribution of individuals affected with autosomal dominant conditions is not uncommon in small families and is almost always due to chance. The husband’s family is an example of how such distortions in the sex ratio of affected relatives often lead to misinformation and mythologies in these families about who is actually at risk for developing the disorder.
When a parent of either sex carries an autosomal dominant mutation, each of his or her children, regardless of sex, has a 50% chance of inheriting that mutant gene. When a large number of families with Huntington disease (or other autosomal dominant disorders) is studied, there is an equal sex distribution of affected individuals.
The husband has a 50% chance of having inherited his mother’s mutant Huntington disease gene.
Both the woman and her husband cared for his mother during her illness and are familiar with the course of the disorder. The woman is now concerned about her husband’s Huntington disease gene status and the possibility that he also will develop symptoms of the disorder. She also wants prenatal testing of the fetal Huntington disease genes although she doubts that pregnancy termination is a consideration. The husband is unsure about whether he wants to learn about his own Huntington disease gene status.
For the husband, due to the current pregnancy, this is an extremely difficult time to learn that he is at 50% risk for a neurodegenerative disorder and may have already transmitted an abnormal gene for that disorder to a child. Whether to have presymptomatic testing for a disorder which currently does not have any effective treatment or prevention requires careful consideration. Abnormal results of such testing may have significant ramifications for psychological well-being and may influence professional, vocational and educational goals, financial planning, and reproductive decisions. Genetic counseling with professionals experienced in this area is strongly recommended.
If prenatal testing by chorionic villus sampling or amniocentesis is undertaken, fetal testing which reveals that the fetus has an abnormal Huntington disease gene will be diagnostic of the husband’s Huntington disease gene status, information that he may not want. In addition, fetal testing is tantamount to testing a child for a condition that usually does not manifest symptoms until adult life and for which there is no current therapy which can prevent or delay onset of symptoms. Professional guidelines strongly discourage testing of minors for such adult-onset conditions. The consensus of these guidelines is that it is preferable to let individuals make autonomous decisions as adults about whether they want this information or not, and that it is preferable to avoid the situation where children might be treated differently by their parents or others if they are known to carry an abnormal gene, even if that gene is unlikely to cause problems for several decades.
The scenario described above raises difficult ethical and legal issues that involve the right of the woman to have information about her fetus, the right of the husband to avoid learning his Huntington disease gene status if he prefers not to know, and the right of the child to make his or her own decision about whether and when to learn of his or her Huntington disease gene status.
For a future pregnancy if the husband does not wish to learn of his Huntington disease gene status but wants to avoid the birth of a child with the abnormal Huntington disease gene, in vitro fertilization with non-disclosing preimplantation genetic diagnosis could be performed. In this scenario, only embryos that are shown to have two normal Huntington disease genes would be implanted in the wife’s womb or frozen for future use, but the couple would not be informed as to whether any abnormal embryos were found.
Further Reading
1. Brinkman RR Mezei MM, Theilmann J et al. (1997) The likelihood of being affected with Huntington disease by a particular age, for a specific CAG size. American Journal of Human Genetics 60 (5):1202–1210.
2. Stevanin G, Fujigasaki H, Lebre AS et al. (2003) Huntington’s disease-like phenotype due to trinucleotide repeat expansions in the TBP and JPH3 genes. Brain 126:1599–1603.
3. Tassicker R, Savulescu J, Skene L et al. (2003) Prenatal diagnosis requests for Huntington’s disease when the father is at risk and does not want to know his genetic status: clinical, legal, and ethical viewpoints. British Medical Journal 326:331–333.
Marfan Syndrome
A 23-year-old woman is referred for genetic counseling at 13 weeks’ gestation due to an increased risk for Down syndrome predicted by first trimester screening. Her family history includes her oldest son who reportedly had hypoplastic left heart syndrome and died at 3 months of age. The woman also reports that her mother, age 58 years, and her maternal grandfather were diagnosed with Marfan syndrome. Both are described as being about 6 feet tall with long arms and legs. Her grandfather died of a ruptured thoracic aortic aneurysm in his 50s. Her mother had repair of a thoracic aortic aneurysm a few years ago. A maternal uncle died of a stroke in his 50s. A maternal cousin died suddenly in his 30s of an unknown cause. The woman believes that she might have been evaluated for Marfan syndrome as a teenager and was not thought to have the disorder.
Because the woman could be affected by Marfan syndrome or another thoracic aortic aneurysm syndrome, echocardiography should be accomplished immediately with particular attention to her aortic root and to her aortic valve. In women with Marfan syndrome who have pre-existing aortic or mitral valve disease or dilatation of the aortic root, the hemodynamic changes during pregnancy significantly increase the risk of aortic dissection and death.
The woman’s echocardiogram is normal and shows only minimal mitral valve prolapse. She was unaware that she could still have Marfan syndrome and have an affected child.
Although Marfan syndrome is a very possible diagnosis in the woman’s mother, given the clinical presentations of the two people who have been labeled with that diagnosis and their dissecting aneurysms, there are other possible diagnoses. Thoracic aortic aneurysm syndromes are inherited in an autosomal dominant manner and some show cystic medial necrosis of the aortic wall, as does Marfan syndrome. Also, at times these aneurysms occur in families where there are aortic valve abnormalities and, rarely, hypoplastic left heart syndrome.
Marfan syndrome, an autosomal dominant disorder of connective tissue, has a wide range of clinical severity and is due to mutations in the FBN1 (fibrillin) gene. Affected organ systems include the eye (myopia and lens dislocation), skeleton (bone overgrowth, scoliosis, and joint laxity), and heart (dilatation of the aorta root, predisposition for aortic dissection, mitral valve prolapse, triscuspid valve prolapse, and enlargement of the proximal pulmonary artery). Some individuals with mild presentations may not be recognized while the severe form can result in early death. The diagnosis is made using clinical criteria and family history. Mutations in the FBN1 gene are also associated with other disorders which have clinical overlap with Marfan syndrome including the MASS phenotype (myopia, mitral valve prolapse, aortic enlargement, and non-specific skin and skeletal features), mitral valve prolapse syndrome, and familial ectopia lentis. In addition, another group of disorders which are not associated with mutations in the FBN1 gene also have clinical similarities to Marfan syndrome. These include familial thoracic aortic aneurysms and aortic dissection (TAAD), vascular (type 4) Ehlers–Danlos syndrome, Loeys–Dietz syndrome, homocystinuria, and others.
Since the woman wants information about her own Marfan syndrome status and risks to her children, an evaluation by a medical geneticist is indicated to see whether her clinical presentation meets the diagnostic criteria for Marfan syndrome. If it does, then sequencing of her FBN1 gene could be performed to look for a disease-causing mutation. Between 70–90% of FBN1 gene mutations are identifiable by gene testing.
The woman has a genetics and ophthalmologic evaluation which does not support the diagnosis of Marfan syndrome.
It is not appropriate to initiate gene sequencing of the woman’s FBN1 genes as her clinical presentation does not meet the diagnostic criteria for Marfan syndrome. Identification of an FBN1 gene mutation in the woman’s mother (presuming that the mother has been correctly diagnosed with Marfan syndrome) would allow definitive exclusion of the woman’s Marfan syndrome status. Clinical evaluation of the mother is warranted as she was diagnosed with Marfan syndrome many years ago, prior to the development of the criteria used to establish a firm clinical diagnosis of the disorder.
The woman’s mother is evaluated by a medical geneticist and does not meet the clinical criteria for Marfan syndrome. However, the family history is strongly suggestive that the thoracic aortic aneurysms have an autosomal dominant basis.
In contrast to Marfan syndrome in which all cases are due to mutations in the FBN1 gene, thoracic aneurysms (ascending and descending) associated with autosomal dominant inheritance show genetic heterogeneity. Also, in contrast to Marfan syndrome where mutations in the FBN1 gene are usually completely penetrant (i.e., almost all carries of an FBN1 gene mutation have manifestations of the disorder), there are families with autosomal dominant inheritance of thoracic aneurysms in which there are unaffected obligate carriers of a gene mutation. In some families, cerebral aneurysms are also present.
Mutations in four genes have been identified to cause thoracic aneurysms. However, sequence analysis of these genes identifies less than 20% of disease-causing mutations. Some families with thoracic aneurysms show linkage to other loci with as yet uncharacterized genes.
Whether the hypoplastic left heart syndrome in the woman’s son was related to thoracic aneurysms affecting other family members is not known and cannot be established. It increases the probability that she is a carrier of a gene which causes thoracic aneurysms. Whether she will develop a thoracic aneurysm (or aneurysms elsewhere) in the future is not known. She should have surveillance for both thoracic and cerebral aneurysms, given the history of a cousin who died of a stroke at a young age. Because she has had a child with hypoplastic left heart syndrome, the risk of another child with congenital heart disease is at least a few percent. Fetal echocardiography is recommended in her pregnancy.
Further Reading
1. Caglayan AO, Dundar M (2009) Inherited diseases and syndromes leading to aortic aneurysms and dissections. European Journal of Cardiothoracic Surgery 35 (6):931–940.
2. Dean JC (2007) Marfan syndrome: clinical diagnosis and management. European Journal of Human Genetics 15 (7):724–733.
3. Dietz H (updated 6/30/2009) Marfan Syndrome. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009.
4. Krischek B, Inoue I (2006) The genetics of intracranial aneurysms. Journal of Human Genetics 51 (7):587–594.
5. Milewicz DM, Guo DC, Tran-Fadulu V et al. (2008) Genetic basis of thoracic aortic aneurysms and dissections: focus on smooth muscle cell contractile dysfunction. Annual Review of Genomics and Human Genetics 9:283–302.
6. Pannu H, Tran-Fadulu V, Milewicz DM (2005) Genetic basis of thoracic aortic aneurysms and aortic dissections. American Journal of Medical Genetics Part C: Seminars in Medical Genetics 139C1):10–16.
Retinoblastoma
A 28-year-old primiparous woman at 10 weeks’ gestation is referred for genetic counseling because she was diagnosed with an advanced unilateral retinoblastoma at age 3 years in another country. At the time of diagnosis, she had metastases in her abdomen. She was treated with radiation and chemotherapy. After enucleation of her eye and completion of her treatment, she experienced no other medical problems. Her family history does not include other relatives with retinoblastoma.
This woman’s history raises three important questions. The first is whether there might be consequences to the fetus from her prior treatment with chemotherapy and radiation. The second is whether there is a hereditary component to her retinoblastoma placing her children at increased risk of recurrence and susceptibility to other malignancies. The third is whether she faces other health consequences based on her having had a retinoblastoma.
Retinoblastoma is a rare childhood cancer. It may arise de novo in an individual or may be the consequence of an inherited mutation. Retinoblastoma is the paradigmatic disorder illustrating the phenomenon of the two-hit model of tumorigenesis.
Retinoblastoma arises when there is homozygosity for an inactivating mutation in the RB1 gene, which is a tumor suppressor gene on the long arm of chromosome 13. One RB1 gene mutation can be inherited from a parent or can arise de novo in a retinoblast during embryogenesis. A heterozygous inactivating mutation in a single RB1 gene in a retinoblast is not sufficient to cause disease; however, it is a prerequisite for disease occurrence. The development of a retinoblastoma requires a “second hit,” a mutation in the homologous RB1 gene, on the other chromosome 13, which occurs as a result of a somatic mutation.
When both the “first hit” and “second hit” in the RB1 gene arise as somatic events in the same retinoblast, the result is a unilateral retinoblastoma. In this case, the individual does not have an increased risk of recurrent retinoblastoma or other malignancies. On the other hand, if the “first hit” in the RB1 gene is due to a germline mutation that has been inherited from a parent, there is an increased risk of multifocal tumors in the same eye, bilateral retinoblastoma due to “second hits” in both eyes and other non-ocular cancers due to “second hits” in the cells of other body tissues. The risk of cancer in other body tissues is correlated with the amount of radiation exposure with heavily irradiated sites having a higher risk, reflecting the higher chance of a radiation-induced “second hit.” Inherited RB1 mutations follow an autosomal dominant pattern of inheritance and cause what is known as the RB1 cancer syndrome.
The woman’s history does not allow us to establish whether she has a sporadic case of retinoblastoma or whether she has a germline mutation and is at risk for transmitting an RB1 gene mutation to her children. In about 15% of individuals with unilateral retinoblastoma who have no family history of the disorder, one of the RB1 gene mutations found in the tumor is also found in DNA obtained from the individual’s peripheral blood. The mutation in the blood cells could be of germline origin inherited from a parent, or present in a mosaic state, having occurred as a somatic mutation after conception.
A pathologic specimen from the woman’s tumor is not available. Sequencing of her RB1 genes is accomplished using DNA obtained from her peripheral blood cells. She is heterozygous for a previously described nucleotide change. DNA testing of her parents does not identify the RB1 gene mutation and examinations of their retinas are normal. The RB1 gene mutation arose due to a new mutation in either the ovum or sperm with which she was conceived, or might have occurred as a somatic mutation early in embryogenesis.
Each of this woman’s children has a 50% chance of inheriting her RB1 gene mutation. Of those with the mutation, 95% will develop a retinoblastoma in at least one eye, and all face a high lifetime risk of developing other cancers. These children need early surveillance by retinoblastoma specialists in ophthalmology and oncology. This woman could also have prenatal diagnosis of the RB1 gene mutation or preimplantation genetic diagnosis for a future pregnancy. For a fetus who is at high risk or known to carry a RB1 gene mutation, ultrasound surveillance is indicated. Some cases of retinoblastoma present during fetal life.
Gonadal function is adversely affected by prior radiation and chemotherapy resulting in increased risks of infertility or decreased fertility of varying degrees depending on the age of treatment, the type and dose of chemotherapy, and whether the pelvis and abdomen were irradiated. For women who become pregnant, the risk of spontaneous abortion is increased, especially among women previously exposed to high-dose radiation.
For women treated in childhood for non-hereditary cancers who are able to become pregnant, the available data indicate the risk of birth defects or childhood cancers in their children does not appear to be increased over that of the general population background risk. These data reflect the experience with older therapeutic agents and may not apply to the effects of newer treatment regimens available today, which include mutagenic drugs other than alkylating agents and may have different mutagenic effects. There are increased risks of prematurity and low birth weight for the offspring of childhood cancer survivors, with the risk primarily among women who have a history of abdominal radiation. This increased risk reflects radiation-induced damage to the uterus.
Because this woman has a significantly increased risk of second malignancies due to her germline RB1 gene mutation, particularly because she was irradiated, careful surveillance for second cancers is indicated. Avoidance of DNA-damaging agents such as tobacco and ultraviolet light may diminish the risk of some RB1-associated cancers.
Further Reading
1. Boice JD Jr, Tawn EJ, Winther JF et al. (2003) Genetic effects of radiotherapy for childhood cancer. Health Physics 85 (1):65–80.
2. Green DM, Sklar CA, Boice JD et al. (2009) Ovarian failure and reproductive outcomes after childhood cancer treatment: results from the Childhood Cancer Survivor Study. Journal of Clinical Oncology 27 (14): 2374–2381.
3. Maat-Kievit JA, Oepkes D, Hartwig NG et al. (1993) A large retinoblastoma detected in a fetus at 21 weeks of gestation. Prenatal Diagnosis 13 (5):377–384.
4. Reulen RC, Zeegers MP, Wallace WHB et al. (2009) Pregnancy Outcomes among Adult Survivors of Childhood Cancer in the British Childhood Cancer Survivor Study. Cancer Epidemiology Biomarkers Prevention 18 (8):2239–2247.
5. Signorello LB, Cohen SS, Bosetti C et al. (2006) Female survivors of childhood cancer: preterm birth and low birth weight among their children. Journal of the National Cancer Institute 98 (20):1453–1461.
6. Winther JF, Boice JD, Frederiksen K et al. (2009) Radiotherapy for childhood cancer and risk for congenital malformations in offspring: a population-based cohort study. Clinical Genetics 75 (1):50–56.
Tuberous Sclerosis
A 23-year-old woman is referred for genetic counseling because two maternal aunts reportedly have Down syndrome. Both aunts are deceased and were institutionalized in their teens in the early 1960s.
A family history of Down syndrome is a frequent reason for referral for genetic counseling. However, the term “Down syndrome” is often used by families as a generic description for mental retardation regardless of the underlying cause. The presence of two second-degree relatives with Down syndrome raises concern that this woman may carry an inherited chromosomal translocation. Even in the absence of chromosomal information on the aunts, she needs a peripheral blood karyotype to determine whether she has an increased risk for having a child with Down syndrome above that predicted by her age. However, if the aunts did not have Down syndrome but rather some other genetic disorder, a normal karyotype for the woman would not have relevance as to whether she is at increased risk for having affected children.
Medical records of the aunts are probably no longer available, and if they were born prior to the mid 1960s, it is doubtful whether chromosomal analysis would have been performed. Descriptions of the aunts and family photographs may be of help in providing more information about their diagnosis.
Relatives provide photographs of her aunts shortly before their institutionalization. Neither appear to have the characteristic facies of Down syndrome; both have disfiguring acne-like lesions on their faces. Upon further questioning of other family members, it was revealed that one of the aunts had a seizure disorder and died of kidney disease in her 30s. The other aunt died of a brain tumor in her 40s.
Based on this information, the diagnosis of Down syndrome in the maternal aunts is extremely unlikely. The aunts likely have a genetic syndrome but establishing their diagnosis with such limited information can be problematic. If they have a single gene disorder, autosomal recessive and autosomal dominant inheritance would be the most likely possibilities. If the former, the risk of occurrence of their disorder in the women’s children would be small, presuming that she is unrelated to her husband. If the latter, the risk could be considerable because autosomal dominant disorders very often have a wide range of clinical expression even among members of the same family. Some affected relatives have such subtle findings that they go undiagnosed. The recognized abnormalities of the aunts (i.e., skin lesions, seizures, mental retardation, brain tumor) could be entered into a genetics database and might help narrow the differential diagnosis. In light of the aunts’ histories, a complete pedigree analysis and genetics evaluation of the woman and her mother are recommended.
The woman and her mother are evaluated by a medical geneticist and the pedigree is expanded. The mother’s first child, an older brother of the woman, was born in another country and died in the first few weeks of life of a rare heart tumor. Both the woman and her mother have normal intelligence and deny any major medical problems. Physical examination of the mother reveals a few hypopigmented macules on her skin. The daughter’s examination also shows hypopigmented macules as well as facial angiofibromas which the woman reports have been present for several years and were previously diagnosed by the woman’s primary care doctor as acneiform lesions for which treatment with retinoic acid was prescribed.
The presence of three or more hypopigmented macules and facial angiofibromas does meet clinical criteria for a definitive diagnosis of tuberous sclerosis complex. The aunts’ clinical descriptions and photographs, and the brother who probably had a cardiac tumor, also support this diagnosis.
Tuberous sclerosis is an autosomal dominant multisystem disorder which displays a high degree of clinical variability within and among families. The disorder is characterized by a number of different dermatologic findings, brain abnormalities including cortical tubers, subependymal nodules, seizures and mental retardation, renal angiomyolipomas and cysts, and cardiac rhabdomyomas and arrhythmias. Central nervous system abnormalities are the leading cause of premature death in tuberous sclerosis. Renal, heart and lung abnormalities may also be associated with significant morbidity and shortened lifespan.
Mental retardation is common in tuberous sclerosis and is present in about half of affected individuals. Central nervous system tumors including cortical tubers and subependymal glial nodules occur in up to 90% of individuals who carry a disease-causing mutation, and 80% have a seizure disorder of variable severity. Almost half of individuals with a disease-causing mutation meet the diagnostic criteria for autistic spectrum disorder.
Based on a clinical diagnosis of tuberous sclerosis, the risk of recurrence in each of the woman’s children is 50%. A recurring theme in genetic counseling of dominant inheritance with variable expression is that predictions about the severity of disease in a child are not possible to make. Given the diagnosis of tuberous sclerosis complex, imaging of the woman’s chest, kidneys, and brain are recommended. Early detection of astrocytomas, renal angiomyolipomas, and lymphangioleiomyomatosis may lead to treatment that ameliorates some of the serious complications of tuberous sclerosis.
The woman is considering pregnancy and wants prenatal diagnosis of tuberous sclerosis.
Tuberous sclerosis can arise from a mutation in either of two genes, TSC1 or TSC2. About 80% of affected individuals who have a family history of the disorder will have an identifiable mutation in one of these genes. Although further locus heterogeneity is not suspected for tuberous sclerosis, not all TSC1 and TSC2 gene mutations can be found in affected individuals due to the size and complexity of these genes and current limitations of the technology. Preimplantation and prenatal genetic diagnosis would both be available to the woman if a disease-causing mutation were found in one of her TSC1 or TSC2 genes.
Further Reading
1. Crino PB, Nathanson KL, Henske EP (2006) The tuberous sclerosis complex. New England Journal of Medicine 355:1345–1356.
2. Northrup H, Kit Sing Au (updated 5/7/2009) Tuberous Sclerosis Complex. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009
3. Schwartz RA, Fernández G, Kotulska K et al. (2007) Tuberous sclerosis complex: advances in diagnosis, genetics, and management. Journal of the American Academy of Dermatology 57 (2):189–202.
4. Verhoef S, Bakker L, Tempelaars AM et al. (1999) High rate of mosaicism in tuberous sclerosis complex. American Journal of Human Genetics 64:1632–1637.
Features of autosomal recessive inheritance
1. The disorder is expressed when the abnormal gene is present on both homologous autosomal chromosomes (homozygosity or compound heterozygosity for an abnormal allele).
2. The risk of recurrence is 25% when both parents are heterozygous carriers.
3. Males and females are equally likely to be affected.
4. Heterozygotes are typically unaffected.
5. Affected individuals are typically restricted to individuals in the same sibship. More than one generation may be affected in consanguineous families or when the condition has a high prevalence in the general population. The rarer the disorder, the more likely the parents are to be consanguineous.
6. Disease expression (phenotype) tends to be similar (“breeds true”) among affected siblings.
7. Unaffected siblings of an affected individual have a 2/3 chance of being heterozygous for the disorder.
Congenital Adrenal Hyperplasia
Case 1 A woman is referred at 6 weeks’ gestation because she has a 3-year-old son who is reported to have congenital adrenal hyperplasia. He was diagnosed at 2 weeks of age after hospitalization for dehydration and vomiting and requires treatment with mineralocorticoids and glucocorticoids. She wants to know the risk of recurrence of her son’s condition and whether prenatal diagnosis can prevent some of the complications of the disorder.
Congenital adrenal hyperplasia (CAH) refers to a group of autosomal recessive disorders associated with varying degrees of adrenal insufficiency and, in some cases, abnormalities of sexual differentiation. These disorders result from defects in genes controlling the synthesis of cortisol from cholesterol by the adrenal cortex. The most common form is 21-hydroxylase deficiency (21-OHD) which accounts for about 95% of cases. Less common forms include lipoid congenital adrenal hyperplasia, 17α-hydroxylase deficiency, 3β-hydroxysteroid dehydrogenase deficiency, and 11β-hydroxylase deficiency.
More specific information is needed about the son’s diagnosis.
Review of the son’s medical record indicates that he has 21-hydroxylase deficiency (21-OHD), diagnosed shortly after birth after a salt-wasting crisis. He has had no further medical problems since appropriate hormonal replacement was initiated and he is developing normally.
The classic form of 21-OHD is associated with severe adrenal insufficiency and very often with salt-losing crises in the first days or weeks of life. In addition, in utero exposure to excessive fetal androgens can lead to varying degrees of genital ambiguity in females due to virilization of the female genitalia. In severe cases, complete labioscrotal fusion and phallic urethra formation occurs. Effects of excessive androgen exposure on subsequent sexual orientation in affected females are also suspected. The classic form of the disorder can also present during the toddler years with early virilization without salt-wasting. In females, the non-classic or mild form of the disorder may be asymptomatic or present with hirsutism and menstrual irregularity during adolescence or with early puberty or sexual precocity in school-age children.
The patient is concerned about the high probability that an affected female fetus will be virilized.
Because 21-OHD is an autosomal recessive disorder, risk of an affected female fetus is 1 in 8 (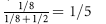 chance that the fetus is female ×
chance that the fetus is female × 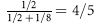 chance that the fetus is affected).
chance that the fetus is affected).
Prenatal diagnosis of 21-OHD can be accomplished by analysis of DNA obtained from chorionic villi or amniocytes if the disease-causing mutations in the causative gene (CYP21A2) have been identified. More than 90% of CYP21A2 gene mutations are identifiable with current laboratory methods. DNA was collected from the patient, her husband and the affected son for mutation testing.
Glucocorticoid (dexamethasone) treatment of the mother can prevent or lessen the severity of virilization of the external genitalia of affected female fetuses, thereby reducing the need for postnatal genital reconstructive surgery. Virilization of the affected female begins as early as 5 menstrual weeks’ gestation. Thus, treatment of the mother should be considered as soon as the pregnancy is recognized. Short-term adverse effects of treatment on maternal health include increased appetite with weight gain, signs of Cushing syndrome, and psychological symptoms. No serious persistent adverse effects on maternal or fetal health have been reported in association with maternal glucocorticoid therapy; however, long-term follow-up studies have not been carried out. Prenatal treatment of the mother with dexamethasone does not eliminate the need for postnatal treatment of an affected child.
Treatment of the mother should be continued until the fetal sex and disease status have been established by chorionic villus sampling. If chorionic villus sampling shows that the fetus is an affected female, treatment of the mother should continue for the duration of the pregnancy. Because the safety of maternal treatment with dexamethasone has not been established and questions have been raised about adverse effects on the mother and/or fetus, treatment should be managed by endocrinologists with expertise in CAH and maternal fetal medicine specialists. Current practice is to discontinue steroid treatment except in the case of an affected female fetus.
The mother is referred to an endocrinologist and placed on dexamethasone therapy pending the results of chorionic villus sampling.
The results of molecular analysis of DNA obtained from peripheral blood leucocytes of the affected son, his mother, and father become available at 9 weeks’ gestation. The affected son is a compound heterozygote for two “severe” mutations causing his classic disease. His father is heterozygous for one of those mutations. His mother carries two mutations, a “severe” mutation which she transmitted to her son, and another “mild” mutation known to be associated with the non-classic disease.
The results of DNA analysis are consistent with the son’s clinical diagnosis of severe disease. An unexpected finding is that the mother also has CAH. Her physical examination is remarkable only for facial hirsutism. She has had no medical or fertility problems.
Because disease-causing mutations have been identified in this family, definitive prenatal diagnosis of CAH is possible by analysis of DNA obtained from uncultured chorionic villi in this and future pregnancies. Preimplantation genetic diagnosis would also be possible for a future pregnancy. The mother should be referred to an endocrinologist for evaluation. Although she may be asymptomatic throughout her entire life, there may be situations in which she may require treatment with glucocorticoids.
The fetus has a 25% risk of inheriting each of the parents’ “severe” mutations. The fetus also has an additional 25% risk of inheriting the father’s “severe” mutation and the mother’s “mild” mutation. If the fetus inherits two “severe” mutations, classic salt-wasting disease as seen in the son would be predicted. However, predictions about the severity of disease will be more difficult to make if the fetus inherits the father’s “severe” mutation and the mother’s “mild” mutation.
In general, compound heterozygotes for a “severe” and a “mild” CYP21A2 mutation usually have clinical expression which is associated with the less severe of the two gene mutations. In the above case, this would predict mild disease as the mother appears to be asymptomatic other than facial hirsutism. However, caution about this conclusion is warranted because the phenotype does not always correlate precisely with the genotype and because other as yet unrecognized genes are also thought to influence clinical manifestations of the disorder.
If chorionic villus sampling reveals that the fetus is female and has inherited two “severe” alleles, dexamethasone treatment of the mother should be continued for the duration of the pregnancy. If the fetus is a compound heterozygote for a “severe” and a “mild” mutation, continued treatment with dexamethasone should be given serious consideration because the possibility of severe disease cannot be excluded. Published experience with the specific mutations in other individuals with the same genotype might help with clinical management in this circumstance.
The accuracy of the results of DNA analysis of chorionic villi is over 99%. Nonetheless, because CAH is a life-threatening disorder in the newborn period, the results of DNA analysis predicting an unaffected fetus should be confirmed immediately after birth by the measurement of serum 17-hydroxyprogesterone. In a baby who is known to be at high risk for CAH based on family history, relying on newborn screening for assessment of disease status is not sufficient.
Case 2 A 29-year-old woman and her husband are seen by a fertility specialist after 3 years of infertility. The woman successfully conceives a pregnancy by in vitro fertilization. As part of the infertility evaluation, the woman is found to have an elevated level of 17-hydroxyprogesterone in her serum and is diagnosed with a mild form of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. She reports menstrual irregularity and acne as a teenager. She has no medical problems. She is referred at 5 weeks’ gestation for consultation regarding the risk of congenital adrenal hyperplasia in the current pregnancy.
The woman may have two “mild” CYP21A2 gene mutations or she may have a “mild” and a “severe” mutation. The chance that her husband is a carrier of a CYP21A2 gene mutation needs to be established in order to determine the risk of an affected fetus.
The incidence of the classic form of congenital adrenal hyperplasia varies according to ethnicity. Among northern Europeans including Eastern European (Ashkenazi) Jews, the incidence is about 1 in 14 000 while among African Americans the incidence is 1 in 42 000. The non-classic form of CAH is one of the most common autosomal recessive disorders with an incidence ranging from 1 in 100 to 1 in 1000 in most populations. An even higher incidence is seen in Hispanics, Ashkenazi Jews, and Mediterraneans. Among Ashkenazi Jews the incidence of the mild form is 1 in 27. Many females and most males with the mild form of CAH go undiagnosed.
The husband is of Ashkenazi Jewish descent.
The chance that the husband carries a “severe” CYP21A2 gene mutation is 1 in 60 [calculated using the Hardy– Weinberg equilibrium (p2 + 2pq + q2 = 1) where q2 = 1 in 14 000], and where p + q = 1 (See page 39 also). The chance that the husband carries at least one “mild” CYP21A2 gene mutation is at least 1 in 3.2 [calculated using the Hardy–Weinberg equilibrium where q2 = 1 in 27]. It is this high because of the high carrier frequency of “mild” mutations among Ashkenazi Jews.
In order to calculate the possibility of classic CAH in the fetus, several possible genotypes of both parents must be considered as there are a number of scenarios that could be present. The mother could have two “mild” CYP21A2 gene mutations, or she could have a “mild” and a “severe” mutation. The father could be a carrier of at least one mild mutation (his chance is 1 in 3.2) or he could be a carrier of a “severe” mutation (his chance is 1 in 60). Although less likely, the father could also have two “mild” mutations or have both a “mild” and a “severe” mutation.
If the mother has two “mild” mutations, the fetus would be an obligate carrier for at least one “mild” mutation. In this case, the risk of the fetus having two “mild” mutations would be 1 in 6.4 [1 (the mother’s chance of transmitting a “mild” mutation) × 1 in 3.2 (the father’s chance of carrying a “mild” mutation) × ½ (the chance that the father transmits his “mild” mutation to the fetus]. The risk of the fetus inheriting a “mild” mutation from the mother and inheriting a “severe” mutation from the father would be 1 in 120 [1 (the mother’s chance of transmitting a “mild” mutation) × 1 in 60 (the father’s chance of carrying a “severe” mutation) × ½ (the chance that the father transmits his “severe” mutation to the fetus)]. In this situation, the fetus could have classic disease although mild CAH would be the more likely outcome. Complete phenotypic predictions before birth are not possible.
If the woman has a “mild” and a “severe” mutation, then there would be a chance of the fetus being homozygous for two “severe” mutations and having the classic form of CAH. This risk would be 1 in 240 [½ (the mother’s chance of transmitting her “severe” mutation) × 1 in 60 (the father’s chance of carrying a “severe” mutation) × ½ (the chance that the father transmits his “severe” mutation to the fetus)].
More than 90% of CYP21A2 gene mutations can be detected by current laboratory methods. Analysis of the parents’ DNA can be accomplished quickly to determine whether there is a risk of classic disease and whether prenatal diagnosis by chorionic villus sampling is indicated. In the meantime, the mother can be treated with dexamethasone to prevent virilization of a female fetus with classic disease.
Prompt evaluation of the newborn by measurement of 17-hydroxyprogesterone should be accomplished in all situations where a parent is known to be affected, even with mild disease.
Further Reading
1. Krone N, Arlt W (2009) Genetics of congenital adrenal hyperplasia. Best Practice and Research Clinical Endocrinology and Metabolism 23 (2):181–192.
2. New M, Nimkarn S (Updated 9/7/2007). 21-Hydroxylase-Deficient Congenital Adrenal Hyperplasia. In: GeneReviews at GeneTests: Medical Genetics Information Resource (database online). Copyright, University of Washington, Seattle. 1997–2009. Available at http://www.genetests.org. Accessed September 2009.
3. Nimkarn S, New MI (2009) Prenatal diagnosis and treatment of congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Molecular and Cellular Endocrinology 300 (1–2):192–196.
4. Speiser PW (2009) Non-classic adrenal hyperplasia. Reviews in Endocrine and Metabolic Disorders 10 (1): 77–82.
Cystic Fibrosis
Case 1 A couple of northern European ancestry comes for genetic counseling to discuss the results of cystic fibrosis gene testing and the risk of the disorder in their current pregnancy. The wife, who is a long-distance runner, is heterozygous for the R117H mutation in the CFTR gene. Her husband, who is also an athlete, is heterozygous for the common ΔF508 mutation. Testing of the couple’s polymorphisms in the intron 8 polythymidine tract of the CFTR gene shows that the mother is homozygous for the 7T allele and the father is heterozygous for the 7T and 9T alleles. The couple has a 4-year-old daughter who is < 5th centile for height and weight, has severe chronic sinusitis, evidence of opacification of the sinus membranes on CT scan, tear duct stenosis bilaterally, and normal motor and cognitive development. She is heterozygous for ΔF508. The couple’s son has normal cognitive and motor development and does not carry either of the parents’ cystic fibrosis gene mutations. Both children have had negative sweat tests. The family history also includes the husband’s brother’s son who also reportedly carries the ΔF508 mutation and had pancreatitis at age 4 years.
The couple has a 25% risk in each pregnancy of a fetus who is a compound heterozygote for ΔF508/R117H. The ΔF508 mutation, which accounts for about 70% of CFTR gene mutations among Caucasians, is a “severe” CFTR mutation and in the homozygous form is usually associated with a classic cystic fibrosis presentation. The R117H mutation (in the absence of a 5T variant in cis) is a “mild” mutation and in combination with ΔF508 on the other homologous chromosome is associated with a wide range of phenotypes including entirely asymptomatic, milder late-onset presentation, or severe disease. Making predictions for this couple is complicated by the information we have about their daughter which is suggestive of a cystic fibrosis-like disorder despite her having inherited only her father’s cystic fibrosis mutation. This raises the possibility that the mother, despite her lack of symptoms, is a compound heterozygote for two cystic fibrosis mutations, only one of which was detected by routine mutation screening. In contrast to mutation screening which only detects common CFTR gene mutations, gene sequencing will find about 99% of CFTR gene mutations.
Cystic fibrosis gene sequencing is indicated for the daughter. In addition to ΔF508, she is found to have a rare cystic fibrosis mutation, L346P, which in other families has been reported in association with mild disease. Further testing of the family showed that the mother also carries this rare mutation and confirms that the daughter is a compound heterozygote for ΔF508/L346P.
It is now possible to explain the discrepancies in the phenotypes of the daughter and father. The father carries only one cystic fibrosis mutation and does not have manifestations of the disorder. In contrast, the daughter has inherited a “severe” mutation from her father and a “mild” mutation from her mother resulting in an intermediate phenotype in which some symptoms of cystic fibrosis are present at an early age. The mother is very unusual in that she carries two “mild” mutations in the CFTR gene and has no apparent symptoms of the disease. As she is a long-distance runner, we can assume she has normal pulmonary function. Whether she is at increased risk for developing later-onset atypical manifestations of cystic fibrosis including bronchiectasis and pancreatitis is not known.
With respect to future children, the couple has a 50% chance of having a child who is a compound heterozygote for two cystic fibrosis mutations (ΔF508/R117H or ΔF508/L346P). While published reports indicate that individuals with the ΔF508/L346P genotype usually have a milder phenotype, definitive predictions for the couple’s children cannot be made.
Case 2 A couple of Ashkenazi Jewish ancestry wants information about the risk of cystic fibrosis in a future pregnancy. The wife had a sister who died at age 18 years of complications of cystic fibrosis. She had severe pulmonary disease and pancreatic insufficiency. The husband has no family history of the disorder. Cystic fibrosis gene mutation screening shows that the husband is heterozygous for the W1282X mutation in the CFTR gene which is the most common CFTR gene mutation seen in Ashkenazi Jews and is usually associated with severe disease. The wife does not carry one of the 25 common cystic fibrosis mutations included in the screening panel. These 25 mutations account for 97% of mutations found in Ashkenazi Jews.
Because the wife had a sister who had cystic fibrosis, the wife’s a priori risk of being a cystic fibrosis mutation carrier is 2/3. The wife’s sister had classic cystic fibrosis making it likely that she had two “severe” mutations which would be included in the common mutation screening panel, but there remains a small chance that the sister had at least one rare mutation which is not detected by routine screening.
Because the wife does not carry a common cystic fibrosis mutation, her calculated residual risk of being a carrier has been reduced to 1 in 18. This Bayesian analysis takes into consideration the woman’s a priori risk of 2/3 and the 97% detection rate in Ashkenazi Jewish individuals for the common mutations which were included in the mutation screening panel. The residual risk of a child affected by cystic fibrosis is 1 in 72 [1 (the chance that the husband is a carrier) × ½ (the chance of transmission of this allele) × 1 in 18 (the chance that the wife is a carrier) × ½ (the chance that she transmits the allele)].
The couple still faces a significant risk of having a child with cystic fibrosis. Although the wife does not carry a common identifiable CFTR gene mutation, there is a small chance that her sister had one (or two) less common cystic fibrosis mutations which were not included in the screening panel and for which the wife was not tested.
In the absence of a living affected family member, there are two approaches that could be used in this situation to provide more information about the wife. She could have gene sequencing which will detect about 99% of cystic fibrosis mutations. A negative result (i.e., no cystic fibrosis mutation detected) would further reduce her carrier risk to about 1 in 50 and the risk of an affected child to about 1 in 200. In the wife’s situation, a better alternative to gene sequencing is available. Her parents, who are obligate carriers of a CFTR gene mutation, are living. Screening them for the presence of common cystic fibrosis mutations has a good chance of providing more information given that their affected daughter had classic disease.
The wife’s parents have cystic fibrosis mutation screening. Her mother is a ΔF508 heterozygote and her father is a W1282X heterozygote.
The results of the wife’s parents’ cystic fibrosis mutation screening tests allow definitive information to be given to the wife. She did not inherit either of her parents’ cystic fibrosis mutations and is almost certainly not a carrier of a CFTR gene mutation. The risk of cystic fibrosis in her children is negligible.
Case 3 A couple is referred for counseling about in vitro fertilization and intracytoplasmic sperm injection. The couple had a 3-year history of infertility. A fertility evaluation revealed the husband has azoospermia due to congenital bilateral absence of the vas deferens. He is otherwise in good health. Subsequent analysis revealed he is heterozygous for ΔF508. His intron 8 polythymidine status is 5T/9T. The wife is heterozygous for the G542X mutation in the CFTR gene which is a classic “severe” mutation and is 7T/7T in her intron 8 polythymidine tract.
Polymorphisms in the intron 8 polythymidine tract of the CFTR gene are known to influence disease expression. The tract may have 5, 7, or 9 thymidines. The 5T variant may be associated with significantly decreased transcription of the functional cystic fibrosis gene. It has clinical importance when it is present in trans with a CFTR gene mutation or another 5T variant (i.e., there is 5T variant on one chromosome 7 and there is a CFTR gene mutation or another 5T tract on the homologous chromosome). Population studies show that the ΔF508 mutation occurs almost exclusively on chromosomes with the 9T variant (i.e., the 9T variant is in cis with ΔF508).
Congenital bilateral absence of the vas deferens (CBAVD) in an otherwise healthy infertile man has a significant chance of being explained by one or more mutations in the CFTR gene. About two-thirds of such men will be compound heterozygotes for two cystic fibrosis mutations or have at least one cystic fibrosis mutation, and almost half will also have the intron 8 5T variant. In contrast, only about 5% of the general population carries a 5T allele. For this reason, cystic fibrosis mutation testing is indicated for all men with CBAVD because they have a significant chance of transmitting a cystic fibrosis mutation to their offspring, which in combination with a mutant maternal allele could result in a more severe phenotype.
In this couple’s pregnancy, the fetus has a 25% chance of being a compound heterozygote for ΔF508/G542X which is associated with severe pulmonary disease and pancreatic insufficiency.
The fetus also has a 25% chance of inheriting G542X from the mother and the intron 8 5T variant from the father. This combination is associated with varying clinical phenotypes ranging from no significant symptomatology to absent vas deferens, chronic or recurrent upper airway disease (bronchi, sinuses, middle ear), asthma, chronic or recurrent pancreatitis, or classic childhood cystic fibrosis. The spectrum of clinical phenotypes is presumably due to differing effects on CFTR transcription from the cystic fibrosis gene on the chromosome that has the 5T variant and other epigenetic or post-translational processes. It is not possible to give predictions about the phenotype aside from frequency observations that suggest that milder phenotypes are more common than severe ones.
Case 4 Cystic fibrosis carrier testing reveals that a woman carries a R117H cystic fibrosis mutation. Her poly T status in intron 8 of the CFTR gene is 5T/7T. Her husband does not carry one of the 97 CFTR gene mutations tested for by a commercial screening panel. His poly T status in intron 8 is 7T/7T. The husband’s ancestry is half Korean and half northern European. The wife has chorionic villus sampling because she is 40 years old and elects to establish the fetal CFTR gene status. Analysis of DNA obtained from uncultured chorionic villi indicates that the fetus inherited the patient’s R117H CFTR mutation. The fetal poly T variant status in intron 8 is 5T/7T. The fetus is heterozygous for both R117H and the 5T variant.
The father had to transmit the 7T allele, thus the results of prenatal diagnosis have also established that the wife’s R117H mutation and her 5T variant in intron 8 are in cis, i.e., on the same chromosome. The presence of the R117H mutation on the same chromosome as the 5T variant results in a severe mutation. If the fetus inherited another cystic fibrosis mutation on the other copy of chromosome number 7 inherited from the husband, this could be associated with severe disease.
The husband is of half-Asian ancestry where the sensitivity of mutation screening for common mutations is decreased compared to individuals of European ancestry. There is still incomplete knowledge of the spectrum of cystic fibrosis mutations and carrier frequencies seen in various Asian populations. The negative results of his cystic fibrosis carrier testing are estimated to reduce the husband’s risk of being a carrier to less than 1 in 100. The residual risk of the cystic fibrosis spectrum of disorders in the current pregnancy is about 1 in 200 although the risk is probably smaller as the calculation conservatively assumes that CFTR mutation screening has a very low mutation detection rate among persons with Asian ancestry.
The option of further cystic fibrosis mutation analysis by DNA sequencing of the husband is a consideration if the family wants more information, because DNA sequencing will find almost all clinically important cystic fibrosis mutations regardless of ancestral background. However, predictions about the severity of symptoms in a child if the husband carries a rare cystic fibrosis mutation may be difficult. Although a mild rather than severe disorder would be more likely, the symptoms of cystic fibrosis that could be present in a child who inherited his mother’s R117H mutation and a rare mutation from his father would include chronic progressive lung disease, pancreatic insufficiency, less severe respiratory and sinus disease, and male infertility. Predictions about the severity of symptoms for any given individual would not be possible to make. However, in this couple’s pregnancy with the information available from prenatal diagnosis, the most likely outcome will be a baby who does not have the cystic fibrosis spectrum of disease.
Further Reading
1. Chillòn M, Casals T, Mercier B et al. (1995) Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. New England Journal of Medicine 332 (22):1475–1480.
2. Cohn JA, Friedman KJ, Noone PG et al. (1998) Relation between mutations of the cystic fibrosis gene and idiopathic pancreatitis. New England Journal of Medicine 339 (10):653–658.
3. Gallati S (2003) Genetics of cystic fibrosis. Seminars in Respiratory and Critical Care Medicine 24 (6):629–638.
4. Kanavakis E, Tzetis M, Antoniadi T et al. (1998) Cystic fibrosis mutation screening in CBAVD patients and men with obstructive azoospermia or severe oligozoospermia. Molecular Human Reproduction 4 (4):333–337.
5. Kerem E (2006) Atypical CF and CF related diseases. Paediatric Respiratory Reviews 7 Suppl 1:S144–146.
6. Lebo RV, Omlor GJ (2007) Targeted extended cystic fibrosis mutation testing on known and at-risk patients and relatives. Genetic Testing 11 (4):427–444.
7. Moskowitz SM, Chmiel JF, Sternen DL et al. (2008) Clinical practice and genetic counseling for cystic fibrosis and CFTR-related disorders. Genetics in Medicine 10 (12):851–868.
8. Rowntree RK, Harris A (2003) The phenotypic conse-quences of CFTR mutations. Annals of Human Genetics 67 (Pt 5):471–485.



