The large majority of Robertsonian translocations are between non-homologous acrocentric chromosomes, and the 13;14 Robertsonian translocation is the most common.
The short arm of an acrocentric chromosome is comprised of the satellite, the satellite stalk, and the proximal short arm. The satellite stalk, also known as the nucleolar organizing region, contains multiple copies of genes coding for ribosomal RNA. An individual with one Robertsonian translocation has only eight satellite stalks instead of the usual ten. However, this reduction is not detrimental although, presumably, a minimum number of stalks with active genes is necessary for normal cellular function.
With rare exceptions, balanced Robertsonian translocations are not associated with adverse effects on health or development. Individuals with balanced Robertsonian translocations do have an increased risk of infertility, recurrent spontaneous abortions, and, depending on the chromosomes involved in the translocation, an increased risk of chromosomally unbalanced viable fetuses and children. These problems occur because the Robertsonian translocation causes abnormal segregation of the chromosomes during meiosis, resulting in gametes with six different possible chromosomal configurations, only two of which are balanced, as shown in Figure 1.1. Thus, at fertilization, the zygote could have an entirely normal chromosomal complement, the balanced translocation, or various combinations of unbalanced products as illustrated in Figure 1.2.
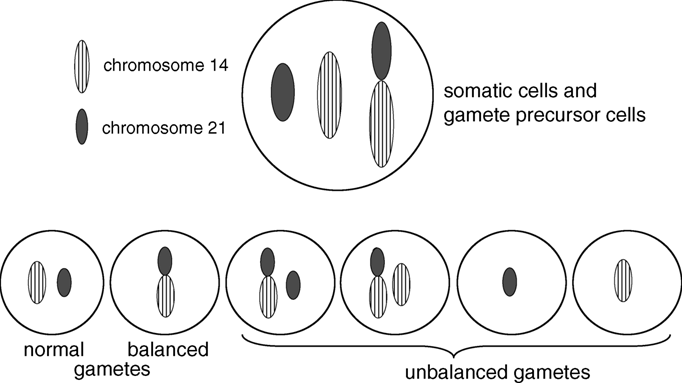
Figure 1.2 Possible chromosomal complements in a zygote after fertilization with a gamete of a non-homologous Robertsonian translocation carrier.
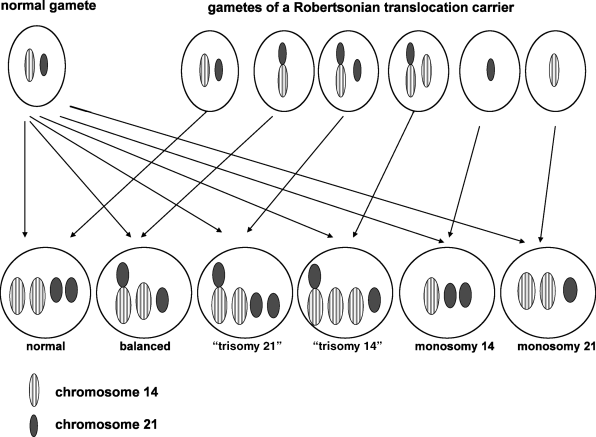
The risk that a balanced Robertsonian translocation carrier will have a viable chromosomally unbalanced fetus at the time of amniocentesis or later in pregnancy is shown in Table 1.2. Although two-thirds of zygotes shown in Figure 1.2 are chromosomally unbalanced, most fetuses of Robertsonian translocation carriers – i.e., all the monosomic fetuses, most fetuses who have three copies of chromosomes 13, and virtually all fetuses with three copies of chromosomes 14, 15, or 22 – will not survive for more than a few days or weeks following fertilization. This accounts for the overall low risk of viable unbalanced fetuses among parents who carry a non-homologous Robertsonian translocation.
Table 1.2 Frequency of unbalanced translocations at the time of amniocentesis* in non-homologous Robertsonian translocation carriers
Data adapted from Gardner and Sutherland (2004) and Silverstein et al. (2002).
| Robertsonian translocation | Balanced carrier mother: % unbalanced at amniocentesis | Balanced carrier father: % unbalanced at amniocentesis |
| 13q14q | 1 | <1 |
| 13q15q | 1 | <1 |
| 13q21q | 10–15 | <1 |
| 13q22q | 1 | <1 |
| 14q15q | 0 | 0 |
| 14q21q | 10–15 | <1 |
| 14q22q | 0 | 0 |
| 15q21q | 10–15 | <1 |
| 15q22q | <1 | <1 |
| *Frequency at livebirth is less than in the second trimester. Estimates for rare Robertsonian translocations are extrapolated from data for the common Robertsonian translocations. Risk of uniparental disomy 14 or 15 is about 0.65% (CI 0.2–2.3%) for Robertsonian translocations involving chromosomes 14 and 15. Carriers of homologous Robertsonian translocations (13q13q, 14q14q, 15q15q, 21q21q, 22q;22q) have a remote chance of having a chromosomally normal conceptus because their gametes will either be nullisomic or disomic for the translocation chromosome. Monosomy for other than the X chromosome results in embryonic death. Trisomy for chromosomes 14 and 15 results in early embryonic death. Fetuses with trisomy for chromosomes 13 or 22 rarely survive into the early second trimester or beyond. 21q21q homologous translocation carriers have a significant risk of a viable fetus with trisomy 21. | ||
The risk of unbalanced embryos surviving into the second trimester or after birth depends on the chromosomes involved in the translocation and which parent carries the Robertsonian translocation. As shown in Table 1.2, the risk is highest for women who carry a non-homologous Robertsonian translocation involving chromosome 21. For 14;21 and 21;22 translocation carriers, the risk at the time of amniocentesis of a viable fetus with Down syndrome is about 15% for a female Robertsonian translocation carrier. In contrast, this risk is only about 1% for a male Robertsonian translocation carrier. The significantly lower risk of viable chromosomally unbalanced fetuses in pregnancies where the father transmits a Robertsonian translocation involving chromosome 21 might reflect selection against chromosomally unbalanced sperm during gametogenesis or fertilization. In contrast to the high risk of Down syndrome in fetuses of women with Robertsonian translocations involving chromosome 21, individuals with non-homologous Robertsonian translocations involving chromosome 13, regardless of their sex, have only a small chance of having a fetus with three copies of chromosome 13 who survives into the second trimester of pregnancy. Individuals with non-homologous Robertsonian translocations involving chromosomes other than 13 and 21 have a very small risk of having a viable fetus with a non-mosaic trisomy even late in the first trimester.
Some individuals with balanced non-homologous Robertsonian translocations experience recurrent early pregnancy loss while others do not. These variations may exist even between members of the same family and do not have a satisfactory explanation. Preimplantation genetic diagnosis might be considered for a couple who have had recurrent miscarriages with unbalanced fetuses.
Due to their rarity, no empiric data are available about the risks of viable unbalanced fetuses for individuals who carry 13;21 and 15;21 Robertsonian translocations. However, we believe the reproductive experience probably would parallel that of the carriers of 14;21 and 21;22 Robertsonian translocations as shown in Table 1.2.
The risk of birth defects in the balanced offspring of a parent who has a balanced Robertsonian translocation involving chromosome 14 (i.e., 13;14; 14;15, 14;21; 14;22), chromosome 15 (i.e., 13;15; 14;15; 15;21; 15;22), and 21;22 is slightly increased over the 3% background risk of birth defects. The slight increase in risk reflects phenomena such as uniparental disomy with imprinting, expression of a recessive gene, or underlying mosaicism in the fetus for a cell line which is trisomic due to an unbalanced Robertsonian translocation.
Uniparental disomy refers to the possibility that the diploid cell line, the “normal” cells, has both of its number 14 or number 15 chromosomes coming from one parent. This arises when early in embryogenesis a trisomic conceptus (trisomy 14 or 15) is “rescued” from embryonic death by a non-disjunction event resulting in loss of one of the number 14 or 15 chromosomes. Both maternal and paternal uniparental disomy 14 and 15 are associated with deleterious effects on normal development. Maternal disomy for chromosome 14 has effects on development of variable severity including prenatal and postnatal growth restriction and facial dysmorphology. Cognitive functioning ranges from normal to severe retardation. Paternal disomy 14 is associated with polyhydramnios, prenatal and postnatal growth restriction, a small thorax, and kyphosis. Maternal disomy 15 is one mechanism that leads to Prader–Willi syndrome and paternal disomy 15 to Angelman syndrome. There is no evidence for deleterious effects of maternal or paternal imprinting of genes on chromosomes 13, 21, and 22 as neither maternal nor paternal uniparental disomy for these chromosomes has been associated with an abnormal phenotype.
In addition to trisomy rescue two other recognized mechanisms, gamete complementation and gamete compensation, can lead to uniparental disomy. Gamete complementation occurs when an ovum missing a specific chromosome is fertilized by a sperm which by chance has two copies of that same chromosome. Gamete compensation refers to the situation where a zygote which is monosomic for a certain chromosome is rescued from embryonic death by a mitotic non-disjunction event leading to duplication of that chromosome.
When a parent carries a Robertsonian translocation involving chromosomes 14 or 15, the risk of uniparental disomy is about 0.65% (95% CI 0.2–2.3%).
Uniparental disomy may cause problems in the fetus due to the presence of recessive alleles or deletions at a gene locus or due to the effects of genetic imprinting. Imprinting refers to the situation in which genes behave differently depending on whether they are inherited from the mother or the father. For example, some genes will only be expressed on a maternally inherited chromosome and others only on a paternally inherited chromosome. For some, but not all genes, absence of a maternal or paternal contribution will result in adverse effects on development or function. Chromosomes containing genes which are imprinted and the clinical consequences of imprinting are listed in Table 1.3.
Table 1.3 Abnormal phenotypes associated with whole chromosome uniparental disomy (UPD)
Adapted from Kotzot D, Utermann G (2005) American Journal of Medical Genetics 136A:287–305. The chromosomes with no clinical descriptions have not been associated with a clinical phenotype at this time.
| Maternal UPD | Chromosome | Paternal UPD |
| 1 | ||
| 2 | ||
| 3 | ||
| 4 | ||
| 5 | ||
| 6 | Transient neonatal diabetes, IUGR, macroglossia | |
| Pre- and postnatal growth retardation; relative macrocephaly; facial dysmorphism resembling Russell–Silver syndrome; developmental delay in some cases, retarded bone age | 7 | |
| 8 | ||
| 9 | ||
| 10 | ||
| 11 | Beckwith–Wiedemann syndrome | |
| 12 | ||
| 13 | ||
| Pre- and postnatal growth retardation; facial dysmorphism (large and broad forehead, high palate, fleshy nasal tip, slight blepharophimosis), muscular hypotonia; precocious puberty; obesity, wide range of intellectual deficits | 14 | Polyhydramnios and premature labor; pre- and postnatal growth retardation; small bell-shaped thorax with short, curved ribs, kyphosis, all of variable severity |
| Prader–Willi syndrome | 15 | Angelman syndrome |
| 16 | ||
| 17 | ||
| 18 | ||
| 19 | ||
| 20 | ||
| 21 | ||
| X | ||
| patXY |
Trisomy rescue during embryogenesis can also lead to the situation in which a fetus is mosaic for a disomic and a chromosomally unbalanced cell line. Phenotypic effects are highly variable and depend on the percentage and distribution of the chromosomally abnormal cells. Chorionic villus sampling or amniocentesis should identify most situations in which the fetus has a large percentage of chromosomally unbalanced cells. Low-level mosaicism, however, may go undetected. The experience with prenatal diagnosis with Robertsonian translocation carriers indicates that if the chorionic villus or amniocyte karyotype is normal, the probability of deleterious underlying mosaicism for a chromosomally unbalanced cell line is very small.
Testing for whole chromosome uniparental disomy 14 or 15 can be accomplished by analysis of DNA obtained from chorionic villi or amniocytes and the peripheral blood cells of the parents. DNA polymorphisms associated with the specific chromosome are analyzed to determine whether there is chromosomal material from both parents. Detection of some cases of uniparental disomy is also becoming available by analysis of the fetal DNA alone.
Case 2 Chromosomal analysis is performed on a newborn after a pediatrician suspects that he has Down syndrome. The karyotype of the baby is 46,XY,-21, + t(21q;21q). The baby has Down syndrome because he has a homologous Robertsonian translocation involving chromosome 21 with two copies of chromosome 21 genes, in addition to a free-standing chromosome 21. Analysis of the parents’ peripheral blood karyotypes is indicated.
Available data show that 90% of unbalanced homologous Robertsonian translocations causing Down syndrome arise as de novo events. In 10% of cases, one of the parents is found to carry the homologous Robertsonian translocation. Homologous translocations for the other acrocentric chromosomes in fetuses are very rare and the percentage of those arising de novo is not known.
If the translocation has arisen de novo in the baby, the risk of recurrence is very small, only slightly increased over the background risk faced by all couples. This slightly increased risk reflects the unlikely possibility that one of the parents has mosaicism for the homologous Robertsonian translocation in his or her germ cells.
If a parent carries the translocation, the chance of having a chromosomally normal child is remote. The gametes of the parent with the homologous Robertsonian translocation contain either two copies of chromosome 21 or no copy. The former situation results in conceptuses with three copies of chromosome 21 and the latter results in conceptuses with monosomy 21 which is lethal during early embryogenesis.
There have been rare reports of chromosomally normal or chromosomally balanced children born to individuals who carry homologous Robertsonian translocations. This can be due to underlying gonadal mosaicism for a normal cell line in the carrier parent, or, by trisomy rescue, gamete compensation or gamete complementation.
Couples in which one member of the couple carries a homologous Robertsonian translocation should be counseled that the chance of a chromosomally balanced fetus is remote. Infertility, recurrent miscarriage, or in the case of individuals with 21;21 and 13;13 Robertsonian translocations, viable fetuses with Down syndrome or trisomy 13 would be expected. Pregnancy could be achieved by either egg or sperm donation.
Further Reading
1. Boue A, Gallano P (1984) Collaborative study of the segregation of inherited chromosome structural rearrangements in 1356 prenatal diagnoses. Prenatal Diagnosis 4:45–67.
2. Cox H, Bullman H, Temple IK (2004) Maternal UPD(14) in the patient with a normal karyotype: clinical report and systematic search for case in samples sent for testing for Prader–Willi syndrome. American Journal of Medical Genetics 127A:21–25.
3. Daniel A, Hook EB, Wulf G (1989) Risks of unbalanced progeny at amniocentesis to carriers of chromosome rearrangements: data from United States and Canadian laboratories. American Journal of Human Genetics 33 (1):14–53.
4. Gardner RJ, Sutherland GR (2004) Chromosome Abnormalities and Genetic Counseling, 3rd edition. Oxford University Press.
5. Kotzot D, Utermann G (2005) Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. American Journal of Medical Genetics 136 (3):287–305.
6. Silverstein S, Lerer I, Sagi M et al. (2002) Uniparental disomy in fetuses diagnosed with balanced Robertsonian translocations: risk estimate. Prenatal Diagnosis 22 (8):649–651.
Chromosomal Mosaicism – Prenatal Diagnosis
Chromosomal mosaicism refers to the situation in which there are cells with different chromosomal complements. It may occur in an individual or in the in vitro culture of cells in a laboratory sample. In liveborns, there is a phenotypic continuum associated with chromosomal mosaicism which ranges from normal or near normal to the classic manifestations of the chromosomal abnormality. Chromosomal mosaicism is often found in amniotic fluid and chorionic villus cell culture. How we determine whether chromosomally abnormal cells found in chorionic villi or amniotic fluid pose a significant risk of underlying mosaicism in fetal tissues will be illustrated by the following cases.
The clinical importance of prenatally diagnosed chromosomal mosaicism depends on a number of factors including the level of mosaicism and the chromosomal abnormality involved.
Three levels of prenatally diagnosed chromosomal mosaicism are defined as follows.
- Level I mosaicism (single cell pseudomosaicism): a single abnormal cell in an otherwise normal study.
- Level II mosaicism (multiple cell pseudomosaicism): the presence of two or more abnormal cells with the same abnormality, which are restricted to a single culture vessel.
- Level III mosaicism (true mosaicism): the presence of abnormal cells with the same abnormality which are found in different culture vessels. True mosaicism means that the abnormal cell line is very unlikely to have arisen in cell culture after chorionic villus sampling or amniocentesis was performed. This conclusion is warranted because the same chromosomal abnormality is present in cells recovered from different culture vessels into which the original, uncultured sample had been placed.
Table 1.4 summarizes relevant information when considering the diagnostic problem of prenatally diagnosed chromosomal mosaicism.
Table 1.4 Prenatally diagnosed chromosomal mosaicism

Mosaicism in Amniotic Fluid for a Common Chromosomal Abnormality (Trisomy 21)
A woman elected amniocentesis based on her age of 35 years. The amniocyte karyotype is 47,XX, + 21/46,XX. Cells with trisomy 21 are recovered from three different cover slips. About 20% of the cells have trisomy 21.
Because cells with a trisomy 21 karyotype were recovered from more than one culture vessel, level III mosaicism (true mosaicism) is present in this study.
Follow-up of pregnancies in which chromosomal mosaicism has been observed in cultured amniocytes suggests that the chromosomal mosaicism may be confined to cells shed from the placenta without phenotypic effects in the fetus. Amniotic fluid cells originate from both the fetus and the placenta although placentally derived cells are a small minority. Nonetheless, abnormal cells recovered in an amniotic fluid sample could be restricted to those originating in the placenta or membranes. Follow-up of pregnancies in which level III (true) mosaicism has been observed in amniotic fluid cells indicate a 70% or higher chance of mosaicism subsequently being confirmed in the fetus, or in the baby after birth. Predictions about the likelihood of demonstrable abnormalities that are associated with the abnormal cells are more difficult to make.
Caution must be exercised when counseling about the overall published concordance rate between amniotic fluid cell mosaicism and the underlying fetal karyotype because the above concordance rate of 70% underestimates the likelihood of a population of abnormal cells in the fetus. Follow-up studies of prenatally diagnosed cases of chromosomal mosaicism have relied mostly on an analysis of peripheral blood cells (and occasionally skin cells) of a liveborn baby or of skin cells from an aborted fetus. This is a major limitation of these studies because mosaicism for rare trisomies (e.g., trisomy 15 – see case below) is often restricted to certain somatic tissues and may be selected against in tissues that are usually submitted for cytogenetic analysis. In addition, the follow-up reports of the liveborns usually discuss the phenotypes present in the newborn period or early infancy. Later-onset developmental problems would not be recognized or reported in the literature. Thus, in the setting of an amniotic fluid study with level III mosaicism for trisomy 21, the likelihood of underlying mosaicism in the fetus is at least 70% and probably higher.
Further testing of the pregnancy to provide more information about the fetal karyotype could be obtained by fetal blood sampling and fetal skin biopsy. In contrast to amniocytes, which may contain a mixture of fetal and placental cells, there is no ambiguity about the origin of cells obtained from fetal blood or skin. If the fetal lymphocyte karyotype is normal, the risk of clinically important mosaicism will be significantly reduced, to a few percent or less. However, normal results of invasive testing cannot completely exclude the possibility of underlying mosaicism in the fetus as the abnormal cell line has a chance of being present in fetal tissues, such as the brain or heart, which are not sampled either before or after birth.
Although some association may exist, correlations between the percentage of abnormal cells recovered in amniocytes or a fetal tissue and the severity of effects on normal development must be made cautiously. It is impossible to know the percentage of abnormal cells in critical body organs.
Detailed ultrasonographic imaging including fetal echocardiography is recommended. The identification of anatomic or growth problems in the fetus, regardless of the results of further invasive testing of the pregnancy, will further raise concerns that the fetus has a significant fraction of abnormal cells. However, normal fetal imaging cannot significantly diminish the risk of clinically underlying mosaicism in the fetus. Even in the non-mosaic form, only about half of fetuses with Down syndrome have detectable signs on ultrasonographic examination. In the setting of mosaicism, in which a normal cell line will moderate or minimize the Down syndrome phenotype, the likelihood of visible anatomic variants or defects is even smaller. It is also important to recognize that a sonographic abnormality might have a totally independent cause unassociated with the mosaicism.
Maternal cell contamination of amniotic fluid is extremely unlikely in the cytogenetics laboratory when the amniocentesis has been uncomplicated and the first few milliliters of fluid are not used for the cytogenetic studies.
When there is no sonographic evidence at the time of amniocentesis of a co-twin, now deceased, who was noted on a first-trimester ultrasonographic examination, cells from the vanishing twin have a very low chance of being present in the amniotic fluid sample.
If the pregnancy continues to term, a karyotype could be obtained from cord blood or from peripheral blood, or from a skin biopsy in the newborn period, or no further testing be performed at all. FISH (fluorescence in situ hybridization) studies of interphase cells such as buccal mucosal cells could also be done. Normal results from testing during the pregnancy and, if desired, of the newborn would provide reasonable inference that the abnormal cell line was unassociated with adverse phenotypic effects in the fetus, but would not completely exclude them.
Mosaicism in Chorionic Villus Cells for a Rare Chromosomal Abnormality (Trisomy 15)
A woman elected chorionic villus sampling based on her age of 40 years. The chorionic villus cell karyotype is 47,XX, + 15/46,XX. Cells with trisomy 15 are found on different cover slips.
Cells with a trisomy 15 karyotype were recovered from two different initial cultures. Level III mosaicism (true mosaicism) is present in this study.
Follow-up of pregnancies in which chromosomal mosaicism has been observed in chorionic villi indicate that the chromosomal mosaicism may be confined to the placenta without phenotypic effects in the fetus. A rough estimate of its presence in the fetus is 40%.
Liveborns with birth defects have been reported who have trisomy 15 mosaicism. Recognition of the entity has been very infrequent, however. The probability of adverse phenotypic effects in the setting of chromosomal mosaicism is related to the percentage and distribution of the cells with abnormal chromosomes. Depending on the percentage and distribution of the abnormal cell line, effects could range from none or mild, to significant abnormalities including mental retardation and structural birth defects. The presence of the abnormal cell line could easily lead to early fetal death.
As with amniotic fluid cell studies, caution must be exercised when counseling about the published overall concordance rate between chorionic villus cell mosaicism and the fetal karyotype. Most studies which attempt to confirm the results of chorionic villi in a liveborn rely only on an analysis of peripheral blood cells and occasionally skin cells. In the past, most studies examined metaphase cells and did not examine interphase cells. More recent studies are including interphase cells. Especially for rare trisomies, e.g., trisomy 15, it is possible that mosaicism for the abnormal cell line is restricted to only certain fetal tissues and may be selected against in tissues that are usually submitted for cytogenetic analysis. The published studies reporting on concordance rates between mosaicism found at the time of chorionic villus sampling and subsequent recovery of abnormal cells in fetal tissue or liveborns are also limited by the fact that relatively little data are available about any specific trisomy.
Further testing of the pregnancy to provide more information about the fetal karyotype could be obtained by amniocentesis, fetal blood sampling, and fetal skin sampling. In addition to standard metaphase analysis of amniocytes, it would be useful to study the amniotic fluid cell population by FISH using a chromosome 15 probe to look for trisomy 15 cells that would not be found by metaphase analysis.
A low risk of adverse phenotypic effects will be predicted if results of amniocentesis are normal, and if detailed ultrasonographic examination documents normal fetal growth and development. However, once mosaicism has been documented in chorionic villi or amniotic fluid, normal results of further imaging or invasive testing cannot completely exclude the possibility of underlying mosaicism in the fetus as the abnormal cell line may be present in fetal tissues that cannot be sampled, and the abnormal cells may not cause an abnormal sonographic phenotype. Trisomy 15 has rarely been recovered from metaphase analysis of peripheral blood lymphocytes, suggesting that these cells may not mature in the marrow or survive in the peripheral circulation very well. In the setting of trisomy 15 mosaicism, normal results of fetal blood sampling would provide limited useful information.
If trisomy 15 cells are found in cultured amniocytes, this substantially increases the likelihood that they are populating fetal tissues. However, amniocytes are derived from both the placenta and the fetus. Recovery of trisomy 15 cells in amniotic fluid does not prove that the fetus has underlying mosaicism as the abnormal cells could still be restricted to the placenta although this subsequent finding would substantially increase the risk of true fetal mosaicism. The overall concordance rate between amniocyte mosaicism and subsequent recovery of the abnormal cells in fetal tissue or a liveborn is suggested to be about 70%. As stated earlier, this is probably an underestimate. It is likely that if trisomy 15 cells are recovered in amniotic fluid, even if ultrasonographic examination reveals normal fetal anatomy, there would be a high probability (>80%) of underlying trisomy 15 mosaicism in the fetus with a wide spectrum of possible effects.
Recovery of trisomy 15 cells from a fetal tissue, e.g., skin or blood, will further raise concerns that the fetus has a significant fraction of abnormal cells. Correlations between the percentage of abnormal cells recovered in amniocytes or a fetal tissue and the severity of effects on normal development must be made cautiously. Particularly for rare trisomies, it may not be appropriate to generalize from the karyotype of fetal lymphocytes or skin fibroblasts to other tissues such as the brain. Also, normal fetal imaging should not be used to conclude that the risk of clinically underlying mosaicism in the fetus is negligible. Karyotypically abnormal cells can have major deleterious effects on organ functioning without gross anatomic defects.
Detailed ultrasonographic imaging including fetal echocardiography is recommended. The identification of anatomic or growth problems in the fetus, regardless of the results of further invasive testing of the pregnancy, will raise concerns considerably that the fetus has suffered phenotypic effects from trisomy 15 mosaicism.
The cells with trisomy 15 could have arisen due to a chromosomal non-disjunction event which occurred in the egg or sperm with which the fetus was conceived. Alternatively, the cells with trisomy 15 could have arisen after conception by a mitotic non-disjunction event. The former explanation for the presence of the trisomy 15 cell line in chorionic villi poses some additional risk of abnormality in the fetus or placenta due to the possibility that the diploid cell line, the “normal” cells, has both of its number 15 chromosomes coming from one parent, a situation known as uniparental disomy. Uniparental disomy arises when a trisomic conceptus, trisomy 15 in this situation, is “rescued” from embryonic death by loss of one of the number 15 chromosomes to generate the mosaic condition with the diploid cell line in addition to the trisomic cell line. If there is uniparental disomy, both members of the homologous chromosome pair in the diploid cell line will have come from the same parent.
Uniparental disomy causes an abnormal phenotype due to imprinting effects or homozygosity at recessive disease loci. Recognizable genetic syndromes have been reported in association with uniparental disomy for chromosome 15 and for uniparental disomy for other chromosomes (see section on Robertsonian translocations). Prader–Willi syndrome occurs when an individual has two chromosome 15s which are maternally inherited. When both chromosome 15s are inherited from a father, this results in Angelman syndrome. Both disorders are associated with significant mental retardation, characteristic physical findings and aberrant behavior. Mosaicism due to trisomy rescue situations arise more frequently, but not exclusively, in the setting of advanced maternal age where there is an increased risk of trisomic conceptuses. The presence or absence of uniparental disomy can be established by analysis of DNA extracted from amniocytes unless there is a major fraction of trisomic cells present. Current uniparental disomy studies often require fetal DNA and DNA from both parents. New methods may make it possible to use fetal DNA only.
Unrecognized maternal cell contamination of cultured chorionic villi is a negligible problem for cytogenetic analysis when chorionic villi are expertly dissected from maternal decidua in an experienced laboratory and the cells are harvested after only a few days in culture.
For twin gestations, it is unlikely that a chorionic villus sample will be contaminated with cells from a living or deceased twin but this possibility cannot be ruled out. The obstetrician’s documentation of the positional relationship of the placentas and the approach of the sampling instrument during the chorionic villus sampling procedure will be helpful in considering the likelihood of contamination.
The trisomy 15 cell line could be confined to the placenta and membranes and still pose problems for the fetus. Placental chromosomal mosaicism may lead to placental dysfunction and poor pregnancy outcome, including intrauterine growth restriction, preterm delivery and intrauterine fetal death. Even if there are no abnormal cells in the fetus, harmful effects could occur because of mosaicism in the placenta.
If the pregnancy continues to term, postnatal chromosome studies could be obtained. If they are normal, and earlier studies of the fetus had been normal, there is a reasonable inference that the abnormal cell line would be unassociated with adverse phenotypic effects in the child, although the evidence would not be conclusive.
Mosaicism in Chorionic Villus Cells for a Different Rare Chromosomal Abnormality (Trisomy 16)
The karyotype of cultured chorionic villi for a 38-year-old woman is 47,XX, + 16. No cell with a normal karyotype was identified. The fetus appeared to have normal growth and development at the time of chorionic villus sampling at 12 weeks’ gestation.
Trisomy 16 is one of the most common chromosomal abnormalities identified in first-trimester spontaneous miscarriages. Identification of a placenta with full or mosaic trisomy 16 is an uncommon finding. Although mosaicism with a normal cell line is not present in this case, only a small fraction of the placenta was sampled.
Liveborns with birth defects due to trisomy 16 mosaicism have been reported. This condition is very rare. Given trisomy 16 cells in the placenta, a rough estimate of its presence in the fetus is 40%. Fetal mosaicism could be associated with a wide range of anatomic and/or functional problems depending on the distribution and number of the trisomy 16 cells. From the limited literature about fetal and placental trisomy 16, up to half of fetuses would either not survive in utero or be abnormal when there is a trisomy 16 placenta. This may be due to placental insufficiency or abnormalities intrinsic to the fetus or both.
Most cases of trisomy 16 mosaicism arise following a trisomy rescue event, increasing the chance of uniparental disomy for trisomy 16. There have been a few reports of chromosomally normal infants with maternal disomy for chromosome 16. Intrauterine growth restriction has been documented in some of these cases. Rarely, birth defects or mental retardation have been present. The scarcity of cases does not allow conclusions as to whether there is a consistent pattern. It is not possible in these cases to determine whether the adverse effects on growth were attributable to imprinting problems associated with uniparental disomy, to dysfunction of a mosaic placenta, or to underlying trisomy 16 mosaicism in tissues which were not analyzed.
Further invasive testing of the pregnancy to provide more information about the fetal karyotype could be obtained by amniocentesis and fetal skin biopsy. Non-invasive testing would include detailed ultrasonographic examination including fetal echocardiography. Low risks of phenotypic effects would be predicted if results of further testing are normal and the fetus appears to have normal anatomy and growth. However, normal results of further testing cannot completely exclude the possibility of underlying mosaicism in the fetus. Trisomy 16 is rarely recovered from metaphase analysis of peripheral blood lymphocytes, suggesting that fetal blood sampling would be unlikely to provide useful information. Future studies of interphase cells in fetal blood using chromosome 16 probes may alter that conclusion, however.
Recovery of trisomy 16 cells from additional invasive testing and/or the identification of anatomic or growth problems in the fetus will considerably raise concerns that the fetus has suffered damaging effects. If growth restriction is the only finding, this might reflect an abnormal placenta with a chromosomally normal fetus. For the latter situation, there would still be uncertainty about long-term deficits for a child. Trisomy 16 cells confined to the placenta are associated with a significant risk of intrauterine growth restriction and perinatal loss.
Mosaicism in Amniotic Fluid for a Rare Chromosomal Abnormality (Trisomy 8)
A 37-year-old woman elects amniocentesis. The karyotype of cultured amniotic fluid cells is 46,XX. One colony contained two cells with a 47,XX, + 8 karyotype. Only cells with normal karyotypes were recovered from a large number of other colonies which were examined.
Two cells with trisomy 8 from a single colony were recovered in an otherwise normal study. This is level II mosaicism (multiple cell pseudomosaicism).
Follow-up of pregnancies in which abnormal amniotic fluid cells are restricted to a single colony or culture vessel do not demonstrate an increased risk of birth defects above the background risk. These studies have significant limitations because they report the experience of level I and level II mosaicism for all chromosomal abnormalities. There is a paucity of follow-up data about mosaicism for specific chromosomes. Thus, the published studies, while reassuring, do not exclude the possibility of a small increased risk of abnormalities associated with underlying mosaicism in the fetus. Furthermore, if detrimental effects on fetal development are present, they may not be recognized at birth. There are numerous anecdotal reports of level I and level II mosaicism being associated with underlying mosaicism in the fetus or liveborn, and, at times, with adverse effects on development.
Especially for rare trisomies, e.g., trisomy 8, it is possible that mosaicism for the abnormal cell line is restricted to only certain fetal tissues and is not present in tissues that are usually submitted for cytogenetic analysis such as blood or skin.
In the setting of only two cells with trisomy 8 in an otherwise normal amniotic fluid cell study, the magnitude of risk is unknown but may be higher than for other trisomies. There are a few case reports of babies diagnosed with trisomy 8 mosaicism syndrome for whom there was only a single cell or even no abnormal cell in amniocyte culture. These reports indicate that trisomy 8 cells may not enter or survive in amniotic fluid as readily as other types of cells. The abnormalities identified in the children with trisomy 8 mosaicism but normal amniocyte karyotypes include mental retardation, cleft palate, and ophthalmologic, cardiac and renal abnormalities. Trisomy 8 mosaicism is also associated with an increased risk of hematopoietic cell malignancies.
Further testing of the pregnancy to determine whether the fetus has trisomy 8 mosaicism is complicated by the knowledge that in affected liveborns the abnormal cell line is often not present in lymphocytes. Therefore, normal results of fetal blood sampling may not be informative with respect to the possibility of underlying trisomy 8 mosaicism although studies of interphase analysis of blood cells by FISH have not been reported. Fetal skin biopsy may have the best chance of detecting trisomy 8 mosaicism if the fetus is truly mosaic. Any further cytogenetic studies should use FISH on interphase cells as well as standard metaphase analysis. If further extensive cytogenetic studies are performed and normal, there may still be a small chance of underlying trisomy 8 mosaicism in the fetus that is higher than for other chromosomes. Abnormal phenotypes have not been identified with uniparental disomy for chromosome 8.
Detailed ultrasonographic imaging including fetal echocardiography is also recommended. However, normal fetal imaging should not be used to conclude that the risk of clinically underlying mosaicism in the fetus is negligible. Karyotypically abnormal cells can have major deleterious effects on organ functioning without gross anatomic defects being present, with the brain being the most vulnerable organ. After birth, chromosome studies can be pursued from both blood and solid tissues.
General Discussion on Prenatally Diagnosed Chromosomal Mosaicism
Further invasive testing of the pregnancy to provide more information about the fetal karyotype exposes the pregnancy to the risks of those procedures. Thus, whether to proceed with further invasive testing must be balanced against the likelihood of finding clinically important results that are related to underlying mosaicism in the fetus. While level III mosaicism for a numerical chromosomal abnormality raises significant concerns about underlying fetal pathology, assessing the risk of abnormalities in a fetus in the setting of level I and level II mosaicism is usually much more problematic. In experienced hands, the risk of pregnancy loss associated with fetal blood sampling and fetal skin sampling is about 1% and 3%, respectively. How to balance these procedure-related risks and the limited information they provide with the uncertain risk of fetal disease is one of the challenges faced by clinicians counseling about prenatally diagnosed mosaicism.
The decision to proceed with further invasive testing of the pregnancy should also be influenced by whether the results, normal or abnormal, will allow revision of prognosis for the fetus. For example, in the setting of level III mosaicism in amniotic fluid for the sex chromosome abnormalities, 47,XXX, 47,XXY, and 47,XYY, recovery of the abnormal cell line in fetal blood would not usually change the uncertainties of prognosis. In the setting of level III mosaicism for 47,XXY (46,XY/47,XXY), the presence of a significant fraction of chromosomally normal cells would be expected to moderate the expression of Klinefelter syndrome and the likelihood for near normal development is increased. However, important aspects of the expression of the symptoms of Klinefelter syndrome depend on the percentage and location of the cells with the extra X chromosome, e.g., brain and testes, information that is not available either prenatally or after birth, regardless of whether 47,XXY cells are recovered in a fetal blood sample.
Mosaicism for a 45,X cell line, either 45,X/46,XX or 45,X/46,XY, has a wide phenotypic spectrum. For 45,X/46,XX mosaicism, there is a phenotypic continuum ranging from the common findings of Turner syndrome to a normal or near normal female. Similarly, males with 45,X/46,XY mosaicism may have varying degrees of ambiguous genitalia or abnormal male genitalia of variable severity, structural abnormalities of the heart and kidneys seen in Turner syndrome, or have a normal or near normal male phenotype. Retrospective data collected about individuals diagnosed postnatally with 45,X/46,XY mosaicism include mainly those individuals who come to medical attention because of structural and/or functional abnormalities and thus cannot be used to provide prognostic information prospectively when the diagnosis of 45,X/46,XY mosaicism is made prenatally.
Predictions about adverse effects on development when 45,X/46,XY mosaicism is present in chorionic villi or amniocytes can be best made by detailed ultrasonographic examination. If fetal anatomy appears normal and the fetus has normally formed male genitalia, there is a high probability (>95%) of a normal-appearing baby at birth although there is a presumed increased risk of abnormal gonadal histology and germ cell tumors. Unfortunately, information about pubertal development, fertility, and risks of malignancy are not yet available for phenotypically normal boys who have been prenatally diagnosed with 45,X/46,XY mosaicism. Because of the possible increased risk of germ cell tumors in this group of boys, careful surveillance is indicated.
For prenatally diagnosed 45,X/46,XX mosaicism, if ultrasonographic examination shows absence of increased nuchal thickening, cystic hygroma, and heart and renal malformations, the risks of lymphatic abnormalities and other structural birth defects often seen in Turner syndrome will be small, but not eliminated. The risks of short stature and ovarian failure are increased but their degree cannot be predicted; there may be gonadal dysgenesis typical of Turner syndrome, less severe effects on ovarian function and fertility, or no apparent effects at all of the 45,X cell line.
In summary, when 45,X/46,XX and 45,X/46,XY mosaicism is detected in chorionic villi or amniotic fluid cells, the recovery of 45,X cells from a fetal tissue is unlikely to add information above that obtained from ultrasonographic examination. Mosaicism for a 45,X cell line can be explored after birth by chromosome studies of peripheral or cord blood cells, skin, including foreskin, or buccal mucosa.
Further Reading
1. Bui TH, Iselius L, Lindsten J (1984) European collaborative study on prenatal diagnosis: mosaicism, pseudomosaicism and single abnormal cells in amniotic fluid cell cultures. Prenatal Diagnosis 4:145–162.
2. Chang HJ, Clark RD, Bachman H (1990) The phenotype of 45,X/46,XY mosaicism: an analysis of 92 prenatally diagnosed cases. American Journal of Human Genetics 46:156–167.
3. de Pater JM, Schuring-Blom GH, Nieste-Otter MA et al. (2000) Trisomy 8 in chorionic villi: unpredictable results in follow-up. Prenatal Diagnosis 20 (5):435–437.
4. European Collaborative Research on Mosaicism in CVS (EUCROMIC) (1999) Trisomy 15 CPM: probable origins, pregnancy outcome and risk of fetal UPD. Prenatal Diagnosis 19 (1):29–35.
5. Hsu LYF, Perlis TE (1983) United States Survey on chromosome mosaicism and pseudomosaicism in prenatal diagnosis. Prenatal Diagnosis 4:97–130.
6. Hsu LY, Yu MT, Neu RL et al. (1997) Rare trisomy mosaicism diagnosed in amniocytes, involving an autosome other than chromosomes 13, 18, 20, and 21: karyotype/phenotype correlations. Prenatal Diagnosis 17 (3):201–242.
7. Johnson A, Wapner RJ, Davis GH et al. (1990) Mosaicism in chorionic villus sampling: an association with poor perinatal outcome. Obstetrics and Gynecology 75:573–577.
8. Kalousek DK, Howard-Peebles PN, Olson SB et al. (1991) Confirmation of CVS mosaicism in term placentae and high frequency of intrauterine growth retardation: association with confined placental mosaicism. Prenatal Diagnosis 11:743–750.
9. Langlois S, Yong PJ, Yong SL et al. (2006) Postnatal follow-up of prenatally diagnosed trisomy 16 mosaicism. Prenatal Diagnosis 26:548–558.
10. Ledbetter DH, Martin AO, Verlinsky Y et al. (1990) Cytogenetic results of chorionic villus sampling: high success rate and diagnostic accuracy in the United States collaborative study. American Journal of Obstetrics and Gynecology 162:495–501.
11. Purvis-Smith SG, Saville T, Manass S et al. (1992) Uniparental disomy 15 resulting from “correction” of an initial trisomy 15. American Journal of Human Genetics 50:1348–1350.
12. van Haelst MM, Van Opstal D, Lindhout D et al. (2001) Management of prenatally diagnosed trisomy 8 mosaicism. Prenatal Diagnosis 21:1075–1078.
13. Vejerslev LO, Mikkelsen M (1989) The European collaborative study of mosaicism in chorionic villus sampling: data from 1986–1987. Prenatal Diagnosis 9:575–588.
14. Worton RG, Stern R (1984) A Canadian collaborative study of mosaicism in amniotic fluid cell cultures. Prenatal Diagnosis 4:131–144.
15. Yong PJ, Barrett IJ, Kalousek DK et al. (2003) Clinical aspects, prenatal diagnosis, and pathogenesis of trisomy 16 mosaicism. Journal of Medical Genetics 40:175–182.



