Neurosurgical Conditions
Brain Tumors
Brain tumors are rare in the first year of life, with an incidence of 1 per 100,000 infants.1 This incidence increases with age. By the age of 2 years, central nervous system (CNS) tumors occur in 2 to 5/100,000 children. Brain tumors represent the most common solid tumors in the pediatric population.2
In the group of brain tumors considered congenital, the most common are teratomas (37%), followed by primitive neuroectodermal tumors (PNETs) (12%), astrocytomas (10–15%), and choroid plexus tumors (8%).3 In a slightly older population of children, teratomas become less frequent and other neoplasms become relatively more common, including astrocytomas (34% of brain tumors in children younger than 24 months of age), PNETs (23%), and ependymomas (11%). There is also a group of tumors that are sometimes considered non-neoplastic. These include developmental tumors, which derive from aberrant proliferative growth during embryonic brain development and include craniopharyngiomas, lipomas, dermoids, and colloid cysts.4
Cerebellar Astrocytomas
Low-grade astrocytomas occur throughout the CNS and constitute a fourth of all pediatric brain tumors. Cerebellar astrocytomas are common (12–17% of pediatric CNS tumors), and their treatment has the most favorable outcome of all intra-axial neoplasms in the CNS.5 Most of these tumors are found in children younger than the age of 10 years. The symptoms of cerebellar astrocytomas include headache (80%), vomiting (80%), gait disturbance, and decreased level of consciousness. Signs include ataxia in 80%, papilledema, cranial nerve palsies (including blindness and diplopia), and dysmetria. The rapid progression of signs and symptoms is often a function of cerebrospinal fluid (CSF) obstruction because the tumor occupies the fourth ventricle or the aqueduct, and causes hydrocephalus. Treatment of the hydrocephalus is often required urgently in very sick children. Temporary diversion of CSF via external ventriculostomies or shunts often precedes removal of the tumor.
Operative excision of these tumors is often possible, but new neurologic deficits can occur postoperatively in 30% of patients, although at least half of these new deficits are transient.6 Postoperative deficits occur because of the proximity of vital brain stem and delicate cranial nerve structures near the tumor, which is often large and firm. Postoperative pseudomeningoceles (the bulging of skin at the occipital incision site) and hydrocephalus are not uncommon (10–25%) and require additional treatment. Temporary continuation of postoperative CSF diversion via external ventriculostomy is favored by many surgeons to reduce the likelihood of permanent ventriculoperitoneal shunts.
Ependymomas
Ependymomas represent about 10% of pediatric brain tumors and are slightly less common than PNETs and astrocytomas. More than two-thirds of ependymomas in children occur in the posterior fossa (Fig. 19-1) and the symptoms they cause are similar to posterior fossa or cerebellar astrocytomas: headache, vomiting, lethargy, and ataxia. These symptoms result from a combination of compression along the dorsal brain stem and hydrocephalus. Many extend from the obex (in the inferior aspect of the fourth ventricle) and then extrude through the lateral opening of this ventricle (foramen of Luschka) into the cerebellopontine angle. Here they invade and compress cranial nerves with resulting facial weakness, diplopia, swallowing dysfunction, and hearing loss. When large, they often extend beyond the foramen magnum and can compress the cervical cord. In regard to their cell of origin, they are not limited to the ependymal layer of the ventricle but can arise from cerebellar, cerebral, or spinal cord parenchyma.
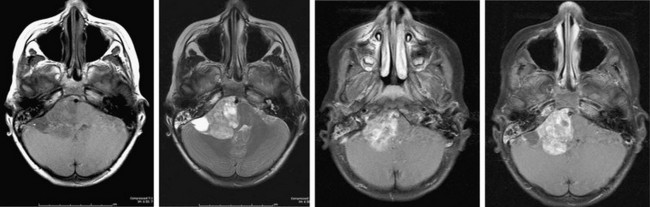
FIGURE 19-1 MR image of a posterior fossa ependymoma. The tumor involves the lateral aspect of the posterior fossa and contains calcifications typical of ependymomas. The brain stem is severely distorted by the mass effect of the tumor. The tumor also compresses cranial nerves on the right side, making complete tumor removal problematic. This patient had right facial weakness, diplopia, and mild left hemiparesis postoperatively.
Aggressive surgical resection has been the goal of treatment of these difficult tumors, but there is high probability of new or worsening neurologic deficits as a result of the operative dissection. There is a correlation between postoperative residual tumor and tumor recurrence and/or progression. In the 30% of children where the tumor is totally resected, there is still a 20% to 40% possibility of recurrence, even after conventional radiotherapy.7 In cases of near-total resection, the rate of progression-free survival falls to 30%.8 Most of the recurrences are local and radiation therapy has become the mainstay of treatment, even with complete resection. Very small children, usually younger than age 3 years, are faced with comparatively greater morbidity from conventional radiotherapy, and radiation therapy is often deferred. Chemotherapy has been used with some success in this group of patients. The role of chemotherapy without irradiation has generally not been favorable in older age groups. Retrospective studies have failed to prove substantial benefit in survival when chemotherapy is added to surgery and radiation therapy for newly diagnosed ependymomas.
Medulloblastomas
Medulloblastomas are the most common malignant solid tumor in children and constitute 20% of pediatric brain tumors.9 They are usually located in the posterior fossa and they are also referred to as PNETs because they are histologically identical to tumors (pineoblastomas, neuroblastomas, and retinoblastomas) located in other locations that are believed to have derived from progenitor subependymal neuroepithelial cells undergoing malignant transformation. Nearly half of medulloblastomas have chromosomal abnormalities, particularly the deletion of 17p chromosome that contains the tumor suppressor gene TP53.10
Hydrocephalus often occurs with medulloblastomas because of their location within the cerebellar vermis (a midline structure), often filling the fourth ventricle (Fig. 19-2). Children with this tumor often have symptoms of elevated intracranial pressure (ICP) (obtundation, headache, nausea/vomiting, irritability) and have signs suggestive of posterior fossa compression (dysmetria, ataxia, diplopia, head tilt, and papilledema). Lumbar puncture should not be done after computed tomography (CT) or magnetic resonance imaging (MRI) has established the presence of a posterior fossa tumor and obstructive hydrocephalus. CSF diversion (usually via an external drain) is usually done either before or in conjunction with craniotomy. Conversion of these temporary devices to permanent ventriculoperitoneal shunts is not uncommon in children with large tumors and marked preoperative ventriculomegaly.
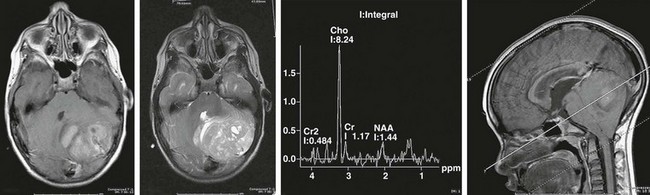
FIGURE 19-2 MRI and MRS (magnetic resonance spectroscopy) of a posterior fossa medulloblastoma, with compression of fourth ventricle. The MRS profile shows a choline peak with depression of N-acetyl aspartate (NAA) typical of aggressive tumors.
Complete resection of medulloblastomas is often possible, although permanent postoperative deficits can occur.6 The ‘posterior fossa syndrome’ can occur postoperatively in 10–15% of children and is characterized by mutism, drooling and swallowing difficulties, ocular palsies, and increasing ataxia.11 These problems resolve entirely in most patients after several months. Improvement is thought to occur with resolution of swelling within the inferior vermis.
Staging is important with medulloblastomas because patients have a predictable outcome depending on age, metastases, pathology, and extent of surgical resection. Poor survival is correlated with age younger than 4 years, residual tumor measuring more than 1.5 cm,2 and tumor dissemination, particularly ‘drop’ metastasis along the spinal column. After craniospinal radiation in eligible patients, survival occurs in 50–70% of patients with standard or low-risk medulloblastomas. Newer treatment protocols include chemotherapy first, which is followed by radiotherapy consisting of lowered cumulative craniospinal doses (24–36 Gy to the entire brain) with hyperfractionated delivery, usually 1 Gy twice daily. Survival in these ‘good risk’ patients is nearly 90% after five years. Survival in high-risk patients is 60–65% with current multimodality therapy, with the worst outcomes in affected infants. Children younger than 3 years of age are usually treated first with chemotherapy, with irradiation deferred for 1 to 2 years. Radiation is sometimes avoided altogether in the 40% with progression-free survival.12
Supratentorial Nonglial and Glial Neoplasms
Ependymomas that occur in the cerebral hemispheres remain problematic in terms of treatment, although hemispheric tumors generally have comparatively better outcomes. Incomplete resection (often because of diffuse involvement within critical brain regions) and leptomeningeal spread are significant adverse risk factors. Age is also a factor. Children younger than 3 years of age have significantly diminished progression-free survival (10–15%) compared with older children.13
As with medulloblastoma, glial and nonglial tumors often have genetic abnormalities, with chromosomal abnormalities and gene mutations. Many low-grade lesions progress to become more malignant. The pathway to this malignant progression is complex. Chromosomal translocations and mutations occur as initiating events before the amplification of deleterious genes that support tumor progression.14–16
With most benign tumors, there exists a strong association between extent of resection and outcome. From the neurosurgical viewpoint, maximal resection should be attempted without inordinate surgical morbidity. The advent of frameless stereotaxy for precise tumor localization, ‘functional’ localization with intraoperative monitoring (e.g., somatosensory evoked potential mapping to determine the location of the motor cortex), presurgical functional MRI to determine location of speech areas, and intraoperative scanning (via real-time ultrasonography [US], CT, or MRI) have each added considerably to the safety of the operation. Nonetheless, the operative approach can only achieve resection of targeted areas. Infiltrative tumors (which typically extend well beyond the target borders) cannot be ablated using current surgical technology. The roles of chemotherapy, molecular manipulation, and conformal radiation therapy remain essential to the goal of controlling high-grade brain neoplasms partially treated with operation. Unfortunately, high-grade brain lesions remain stubbornly resistant to the intensive multimodality treatments that follow surgical resection. Survival curves for highly malignant brain lesions have not changed substantially in the past several decades.
Radiotherapy for Pediatric CNS Tumors
The target for radiation therapy is cellular DNA. Ionizing radiation damages double-stranded DNA, leading to cell death. Unlike normal cells, which have a preserved ability to repair radiation damage, neoplastic cells often are replicating at abnormally high rates and radiation interferes with their mitotic or proliferative ability. With slowly growing tumors such as craniopharyngiomas, the response to radiation is subtle and such tumors may take many months to show a clinical response. The critical sublethal dose required to preserve normal tissue but damage brain tumors, the so-called therapeutic window, is quite well understood and depends on a number of factors, including vulnerability of affected tissue (which can depend on the age of the patient and locale of the target; optic nerve radiation, as an example, is poorly tolerated), tumor vulnerability, volume irradiated, total dose, fraction size, and interfraction interval. As total volume of irradiation increases, the cumulative radiation dose must necessarily decrease to reduce the morbidity of treatment. The conventional cumulative radiation dose for most pediatric CNS tumors is in the 50–60 Gy range, although some tumors (e.g., germinomas) are much more sensitive to radiation and can respond to treatment in the 30–50 Gy range.17 Tumor type is therefore an important determinant of the effectiveness of radiation therapy, and biopsy is often a prerequisite for treatment.
Treatment of Spasticity and Movement Disorders
Spasticity occurs because of an imbalance of excitatory Ia afferent nerves from muscle spindles into the spinal cord and inhibitory descending impulses from the basal ganglia and cerebellum. In most children, the inhibitory impulses are diminished because of early CNS injury or injury to the spinal cord, which conducts the descending inhibitory impulses. Hence, treatment is directed toward either increasing the inhibitory neurotransmitters (usually γ-aminobutyric acid [GABA]) or reducing the afferent excitatory transmission from muscle spindles. Baclofen achieves the former, and dorsal rhizotomy (via cutting afferent nerve roots) interrupts the reflex transmission from muscle spindles.18,19
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


