Chapter 589 Neurocutaneous Syndromes
589.1 Neurofibromatosis
Clinical Manifestations and Diagnosis
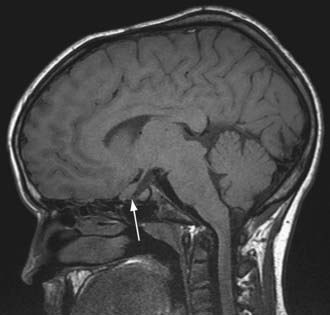
NF-1 is the most prevalent type, with an incidence of 1/3,000, and is diagnosed when any 2 of the following 7 features are present: (1) Six or more café-au-lait macules over 5 mm in greatest diameter in prepubertal individuals and over 15 mm in greatest diameter in postpubertal individuals. Café-au-lait spots are the hallmark of neurofibromatosis and are present in almost 100% of patients. They are present at birth but increase in size, number, and pigmentation, especially during the first few yrs of life (Fig. 589-1). The spots are scattered over the body surface, with predilection for the trunk and extremities but sparing the face. (2) Axillary or inguinal freckling consisting of multiple hyperpigmented areas 2-3 mm in diameter. Skinfold freckling usually appears between 3 and 5 yr of age. The frequency of axillary and inguinal freckling has been reported to be greater than 80% by 6 yr of age. (3) Two or more iris Lisch nodules. Lisch nodules are hamartomas located within the iris and are best identified by a slit-lamp examination (Fig. 589-2). They are present in >74% of patients with NF-1 but are not a component of NF-2. The prevalence of Lisch nodules increases with age, from only 5% of children <3 yr of age, to 42% among children 3-4 yr of age, and virtually 100% of adults ≥21 yr of age. (4) Two or more neurofibromas or 1 plexiform neurofibroma. Neurofibromas typically involve the skin, but they may be situated along peripheral nerves and blood vessels and within viscera including the gastrointestinal tract. These lesions appear characteristically during adolescence or pregnancy, suggesting a hormonal influence. They are usually small, rubbery lesions with a slight purplish discoloration of the overlying skin. Plexiform neurofibromas are usually evident at birth and result from diffuse thickening of nerve trunks that are frequently located in the orbital or temporal region of the face. The skin overlying a plexiform neurofibroma may be hyperpigmented to a greater degree than a café-au-lait spot. Plexiform neurofibromas may produce overgrowth of an extremity and a deformity of the corresponding bone. (5) A distinctive osseous lesion such as sphenoid dysplasia (which may cause pulsating exophthalmos) or cortical thinning of long bones (e.g., of the tibia) with or without pseudoarthrosis. (6) Optic gliomas are present in approximately 15% of patients with NF-1 and represent mostly low-grade astrocytomas. They are the main CNS tumor with a marked increased frequency in NF-1. Because of their growth, it is recommended that all children age 10 yr or younger with NF-1 undergo annual ophthalmologic examinations. When they progress, visual symptoms are produced because the tumors enlarge and put pressure on the optic nerves and chiasm resulting in impaired visual acuity and visual fields. Extension into the hypothalamus can lead to endocrine deficiencies or failure to thrive. The MRI findings of an optic glioma include diffuse thickening, localized enlargement, or a distinct focal mass originating from the optic nerve or chiasm (Fig. 589-3). (7) A first-degree relative with NF-1 whose diagnosis was based on the aforementioned criteria.
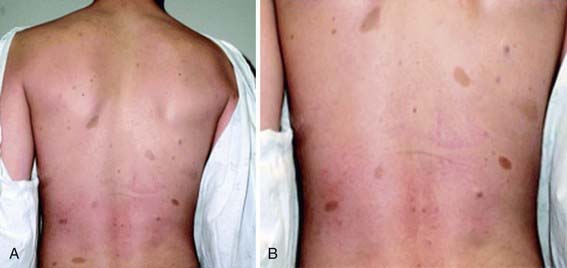
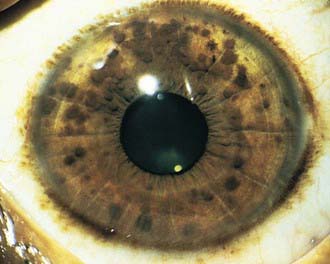
Figure 589-2 Neurofibromatosis 1. Pigmented hamartomas of the iris (Lisch nodules).
(From Zitelli BJ, Davis HW: Atlas of pediatric physical diagnosis, ed 4, St Louis, 2002, Mosby, p 507.)
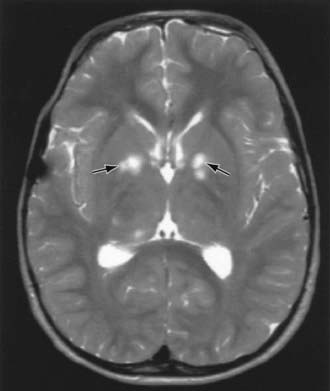
Children with NF-1 are susceptible to neurologic complications. MRI studies of selected children have shown abnormal hyperintense T2 weighted signals in the optic tracts, brainstem, globus pallidus, thalamus, internal capsule, and cerebellum (Fig. 589-4). These signals, “unidentified bright objects (UBOs),” tend to disappear with age; most have disappeared by 30 yr of age. It is unclear what the UBOs represent pathologically, and there is disagreement as to the presence and number of UBOs and their relationship to learning disabilities, attention deficit disorders, behavioral and psychosocial problems, and abnormalities of speech among affected children. Therefore, imaging studies such as brain MRIs should be reserved only for patients with clinical symptoms.
One of the most common complications is learning disability affecting approximately 30% of NF-1 children. Seizures are observed in approximately 8% of NF-1 patients. The cerebral vessels may develop aneurysms, or stenosis resulting in moyamoya disease (Chapter 594.1). Neurologic sequelae of these vascular abnormalities include transient cerebrovascular ischemic attacks, hemiparesis, and cognitive defects. Precocious puberty may become evident in the presence or absence of lesions of the optic chiasm and hypothalamus. Malignant neoplasms are also a significant problem in patients with NF-1, affecting approximately 3% of patients. A neurofibroma occasionally differentiates into a malignant peripheral nerve sheath tumor (MPNST). The incidence of pheochromocytoma, rhabdomyosarcoma, leukemia, and Wilms tumor is higher than in the general population. Scoliosis is a common complication found in about 10% of the patients. Patients with NF-1 are at risk for hypertension, which may result from renal vascular stenosis or a pheochromocytoma.
NF-2 is a rarer condition, with an incidence of 1/25,000, and may be diagnosed when 1 of the following 4 features is present: (1) bilateral vestibular schwannomas; (2) a parent, sibling, or child with NF-2 and either unilateral vestibular schwannoma or any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, or posterior subcapsular lenticular opacities; (3) unilateral vestibular schwannoma and any 2 of the following: meningioma, schwannoma, glioma, neurofibroma, or posterior subcapsular lenticular opacities; (4) multiple meningiomas (2 or more) and unilateral vestibular schwannoma or any 2 of the following: schwannoma, glioma, neurofibroma, or cataract. Symptoms of tinnitus, hearing loss, facial weakness, headache, or unsteadiness may appear during childhood, although signs of a cerebellopontine angle mass are more commonly present in the 2nd and 3rd decades of life. Although café-au-lait spots and skin neurofibromas are classic findings in NF-1, they are much less common in NF-2. Posterior subcapsular lens opacities are identified in about 50% of patients with NF-2. The NF2 gene (also known as merlin or schwannomin) is located on chromosome 22q1.11. The frequency of lesions associated with NF-2 is noted in Table 589-1.
Table 589-1 FREQUENCY OF LESIONS ASSOCIATED WITH NEUROFIBROMATOSIS TYPE 2
| FREQUENCY OF ASSOCIATION WITH NF2 | |
|---|---|
| NEUROLOGIC LESIONS | |
| Bilateral vestibular schwannomas | 90-95% |
| Other cranial nerve schwannomas | 24-51% |
| Intracranial meningiomas | 45-58% |
| Spinal tumors | 63-90% |
| Extramedullary | 55-90% |
| Intramedullary | 18-53% |
| Peripheral neuropathy | Up to 66% |
| OPHTHALMOLOGIC LESIONS | |
| Cataracts | 60-81% |
| Epiretinal membranes | 12-40% |
| Retinal hamartomas | 6-22% |
| CUTANEOUS LESIONS | |
| Skin tumors | 59-68% |
| Skin plaques | 41-48% |
| Subcutaneous tumors | 43-48% |
| Intradermal tumors | Rare |
From Asthagiri AR, Parry DM, Butman JA, et al: Neurofibromatosis type 2, Lancet 373:1974–1984, 2009, Table 1.)
American Academy of Pediatrics Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics. 1995;96:368-372.
Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet. 2009;373:1974-1984.
Baser ME, R Evans DG, Gutmann DH. Neurofibromatosis 2. Curr Opin Neurol. 2003;16(1):27-33.
Cnossen MH, de Goede-Bolder A, van den Broek KM, et al. A prospective 10 year follow up study of patients with neurofibromatosis type 1. Arch Dis Child. 1998;78:408-412.
DeBella K, Szudek J, Friedman JM. Use of the National Institutes of Health criteria for diagnosis of neurofibromatosis 1 in children. Pediatrics Mar. 2000;105:608-614.
Evans DGR. Neurofibromatosis type 2 (NF2): a clinical and molecular review. Orphanet J Rare Dis. 2009;4:16.
Hersh JH, Committee on Genetics. Health supervision for children with neurofibromatosis. Pediatrics. 2008;121:633-642.
Hofman KJ, Harris EL, Bryan RN, et al. Neurofibromatosis type 1: the cognitive phenotype. J Pediatr. 1994;124:S1-S8.
Krab LC, Aarsen FK, de Goede-Bolder A, et al. Impact of neurofibromatosis type 1 on school performance. J Child Neurol. 2008;23:1002-1010.
Messiaen L, Yao S, Brems H, et al. Clinical and mutational spectrum of neurofibromatosis type 1-like syndromes. JAMA. 2009;302:2111-2118.
Plotkin SR, Stemmer-Rachamimov AO, Barker FGII, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361:358-367.
Rizzo JF, Lessell S. Cerebrovascular abnormalities in neurofibromatosis type 1. Neurology. 1994;44:1000-1002.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


