Chapter 492 Neuroblastoma
Pathogenesis
The etiology of neuroblastoma in most cases remains unknown. Familial neuroblastoma accounts for 1-2% of all cases, is associated with a younger age at diagnosis, and has been linked to mutations in the Phox2B and ALK genes. Neuroblastoma is associated with other neural crest disorders, including Hirschsprung disease, central hypoventilation syndrome, and neurofibromatosis type I, and potentially congenital cardiovascular malformations (Table 492-1). Children with Beckwith-Wiedemann syndrome and hemihypertrophy also have a higher incidence of neuroblastoma. Increased incidence of neuroblastoma is associated with some maternal and paternal occupational chemical exposures, farming, and work related to electronics, although no single environmental exposure has been shown to cause neuroblastomas.
Table 492-1 SYNDROMES ASSOCIATED WITH NEUROBLASTOMA
| EPONYM | FEATURES |
|---|---|
| Pepper syndrome | Massive involvement of the liver with metastatic disease with or without respiratory distress. |
| Horner syndrome | Unilateral ptosis, myosis, and anhidrosis associated with a thoracic or cervical primary tumor. Symptoms do not resolve with tumor resection. |
| Hutchinson syndrome | Limping and irritability in young child associated with bone and bone marrow metastases. |
| Opsoclonus-myoclonus-ataxia syndrome | Myoclonic jerking and random eye movement with or without cerebellar ataxia. Often associated with a biologically favorable and differentiated tumor. The condition is likely immune mediated, may not resolve with tumor removal, and often exhibits progressive neuropsychological sequelae. |
| Kerner-Morrison syndrome | Intractable secretory diarrhea due to tumor secretion of vasointestinal peptides. Tumors are generally biologically favorable. |
| Neurocristopathy syndrome | Neuroblastoma associated with other neural crest disorders, including congenital hypoventilation syndrome or Hirschsprung disease. Germline mutations in the paired homeobox gene PHOX2B have been identified in a subset of patients with this disease. |
From Park JR, Eggert A, Caron H: Neuroblastoma: biology, prognosis, and treatment, Pediatr Clin North Am 55:97-120, 2008.
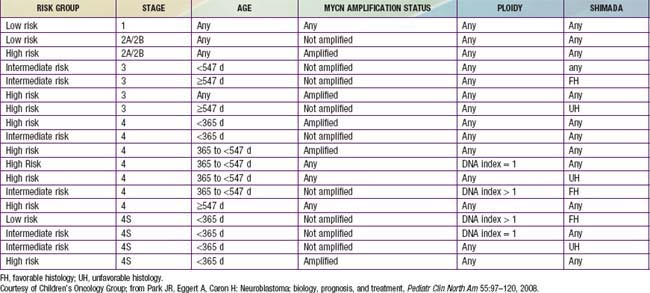
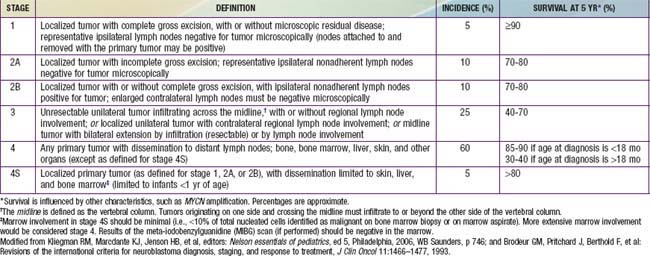
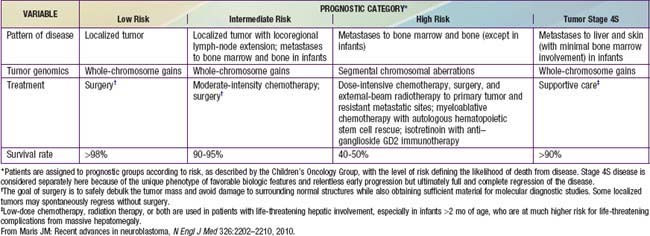
Genetic characteristics of neuroblastoma tumor tissue that are of prognostic importance include amplification of the MYCN (N-myc) proto-oncogene and tumor cell DNA content, or ploidy (Tables 492-2 to 492-4). Amplification of MYCN is strongly associated with advanced tumor stage and poor outcomes. Hyperdiploidy confers better prognosis if the child is <1 yr of age at diagnosis. Other chromosomal abnormalities, including loss of heterozygosity (LOH) of 1p, 11q, and 14q, and gain of 17q, are commonly found in neuroblastoma tumors and are associated with worse outcomes. In addition, many other biologic factors have been shown to be associated with neuroblastoma outcomes, including tumor histology and vascularity and the expression levels of nerve growth factor receptor (TrkA, TrkB), ferritin, lactate dehydrogenase, ganglioside GD2, neuropeptide Y, chromogranin A, CD44, multidrug resistance–associated protein, and telomerase. These factors and many others are under investigation in clinical trials to determine whether they can be used to reduce therapy for children predicted to fare well with minimal therapy and to intensify therapy for those predicted to be at high risk for relapse.