17.4 Neural tube defects, large heads and hydrocephalus
Neural tube defects
Clinical features
NTDs may be classified as in Box 17.4.1.
Box 17.4.1 Classification of neural tube defects
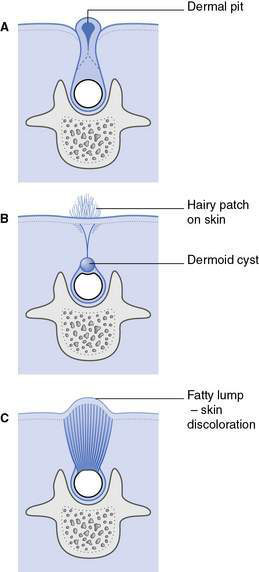
Spina bifida occulta (Fig.17.4.1)
• One or more vertebral arches are incomplete posteriorly but the overlying skin is intact
• Diagnosed incidentally, for example as the result of X-ray of the spine
• Spinal cord usually normal; however, abnormalities of the spinal cord can occur
• Ectodermal abnormalities may be associated with pigmented naevus, angioma, hirsute patch, dimple or dermal sinus on overlying skin
• The ectodermal component may communicate with the dura; may pose some risk of intraspinal infection (if associated with a dural sinus)
Spina bifida cystica
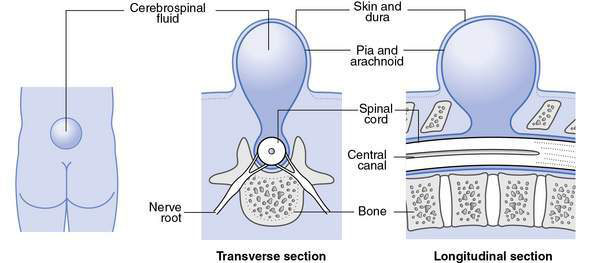
• Meningocele (Fig. 17.4.2), where the spinal cord is not involved:
• Herniation consists of meningeal cyst filled with cerebrospinal fluid, in absence of other malformations, neurological signs are normal
• Not associated with hydrocephalus
• Constitute less than 10% of all cases of spina bifida cystica
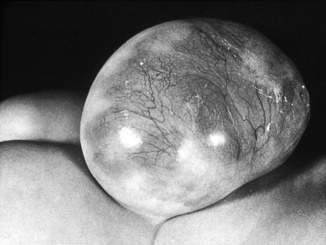
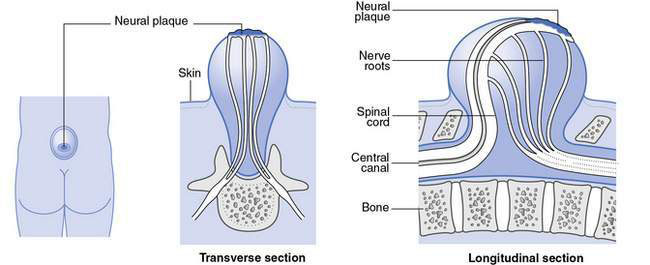
• Myelomeningocele (Figs 17.4.3 & 17.4.4), in which vertebral column skin, meninges and spinal cord are involved:
• Almost always obvious at birth
• May occur anywhere along the length of the spinal column, with lumbar and lumbosacral regions being the most frequent anatomical levels
• Abnormal spinal cord tissue and nerve roots may be readily apparent macroscopically
• There may be spinal abnormalities such as kyphosis at the site of the lesion
• Functional deficits almost always include:
Management of myelomeningocele
• to promote good health in the short and long term
• to promote maximum function in the child so that, as nearly as possible, normal developmental sequences and timing can be followed to enable maximal independence for the child and family
• to support good family functioning to assist the child to reach their maximum potential within the family and community.
Specific problems in the management of the newborn with spina bifida
1. Children with high lesions (thoracic and thoracolumbar), significant hydrocephalus at birth, major kyphosis or other significant problems (either congenital or acquired) have a significantly increased mortality rate in early life and substantial morbidity if they survive. In these circumstances, in discussion with the family, supportive care only may be recommended. If the infant survives the perinatal period, elective surgical care may be indicated. In the absence of such adverse factors, in discussion with the family, early surgical repair/removal of the lesion usually is recommended.
2. Careful serial evaluation of head circumference and ventricular size by ultrasonography or computed tomography (CT) will indicate whether hydrocephalus is developing. Once it is established that progressive hydrocephalus is present, a shunt procedure is recommended.
3. Baseline orthopaedic, urological and neurosurgical assessments provide the basis for ongoing discussions with the family and management of the condition. Occasionally, active urological intervention is required for urinary retention.
4. It is critical to begin to establish an empathetic, therapeutic relationship with the parents in the newborn period that forms the foundation for ongoing support throughout childhood.



