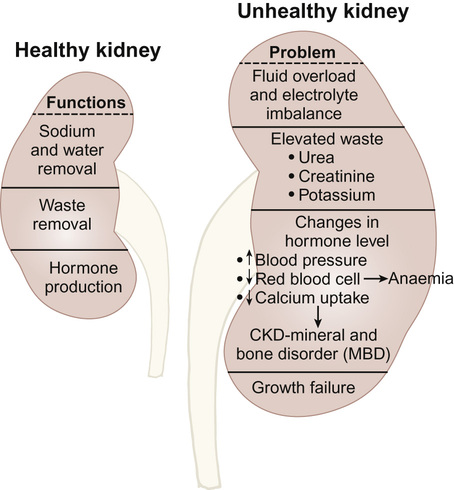
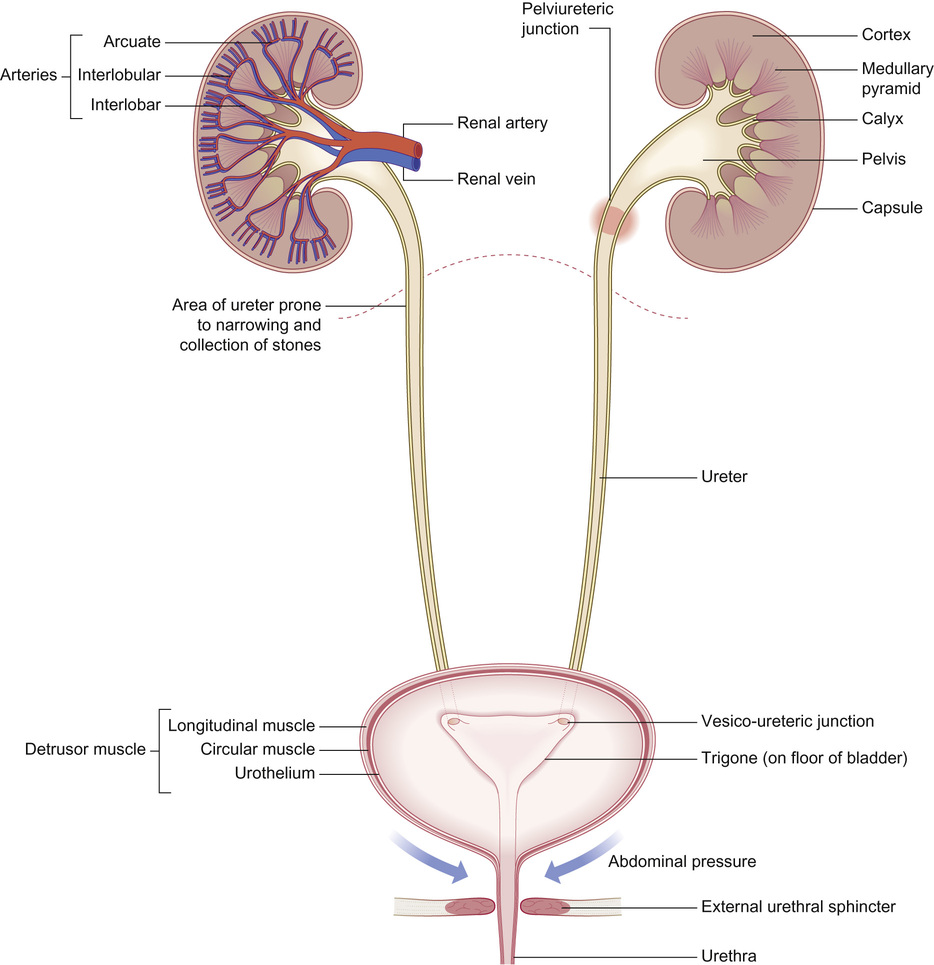
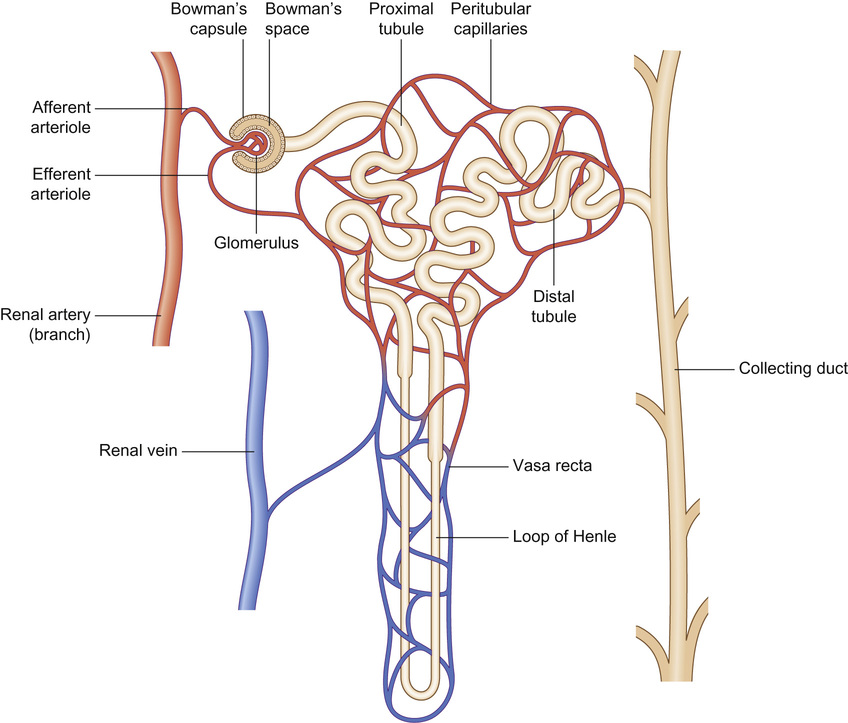
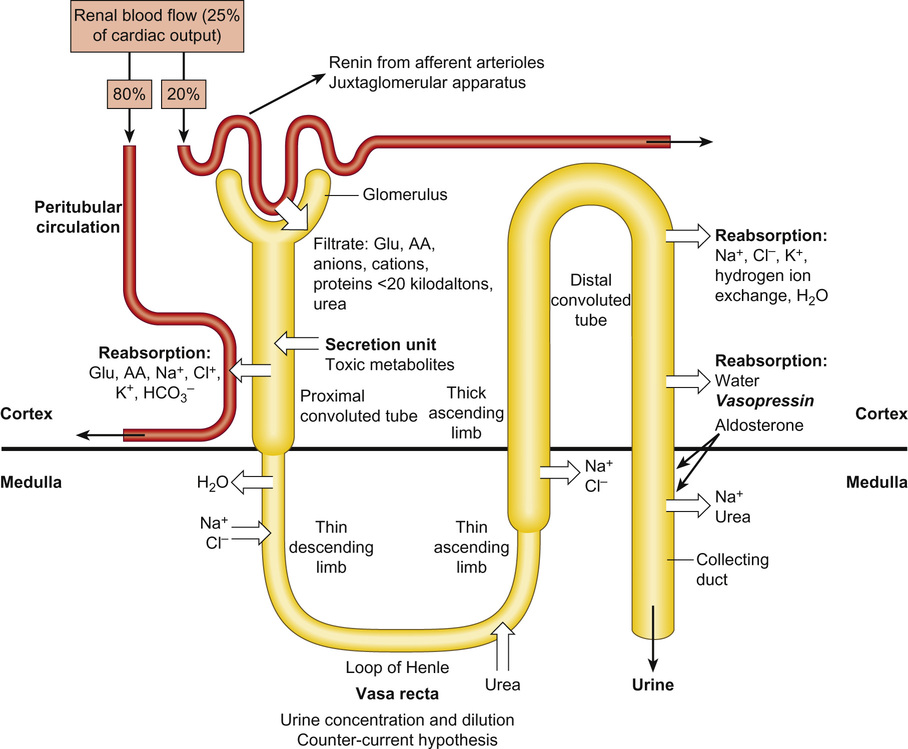
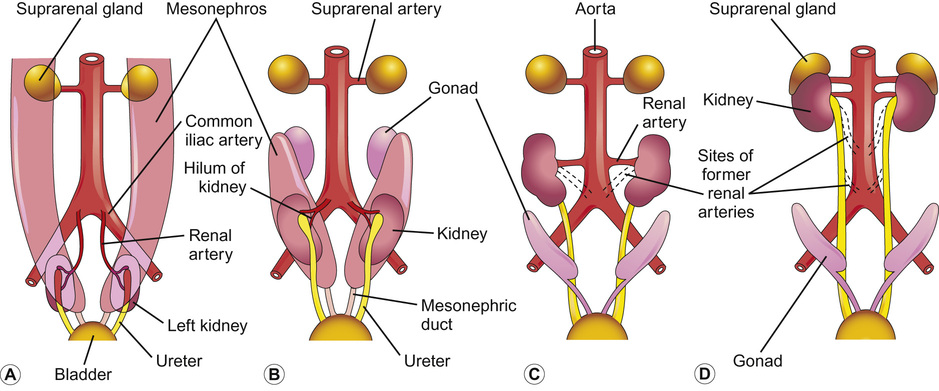
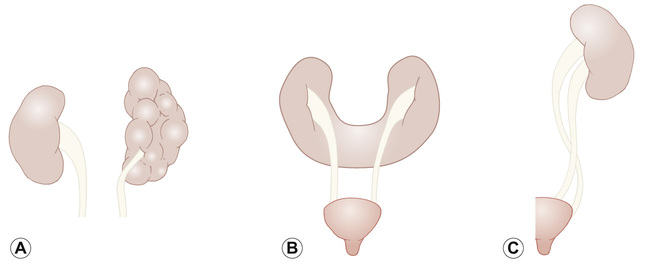
Rajiv Sinha, Stephen D Marks • Understand the critical anatomy and embryology of the urinary tract • Understand the relevant physiology of the kidney and understand how dysfunction relates to disease • Know the commoner presentations of renal disease in children • Have a logical rationale for the investigation of renal disease and disorders of the urinary tract • Understand the principles of water balance and rehydration The kidneys and urinary tract are a critical organ system in enabling homeostasis. Their principle functions are those of water, electrolyte and acid–base balance. They also play an important role in hormone secretion, red cell production and the control of blood pressure. The functions of the renal tract are most clearly elucidated by examining the results of renal failure and renal disease (Fig. 19.1). Kidneys are retroperitoneal structures located in the paravertebral space; the right kidney is slightly lower than the left. At birth, average length of the kidney is 4.5–5.5 cm, whereas an adult kidney measures 10–11.5 cm in length and 5–7 cm in width. The anterior surface of the kidney lies in contact with the duodenum on the right side and the pancreas on the left side. Variable portions of colon may be in contact with the inferior pole of the anterior surface of the kidneys, and on the left side the spleen wraps the anterolateral aspects of the upper half of the kidney. The 12th rib and a portion of the 11th rib cover the upper third of the posterior surface of the left kidney, whereas the 12th rib may touch the upper pole of the right kidney. The outer surface of the kidney is covered by a thin but firm capsule. The anatomical position of the kidney explains the retroperitoneal approach for renal biopsy and the choice of lower pole of the right kidney. A longitudinal cut section of the kidney and its relationship to the other structures in the urinary tract is shown in Figure 19.2. Nephrons (Fig. 19.3) are the ultrastructural units of the kidney, of which we have around a million in each kidney. The nephron comprises a glomerulus connected to a tubule which in turn drains into a collecting duct; ultimately, they join and drain into the calyces at the renal pyramids. The glomerulus generates filtrate through ultrafiltration of blood brought in by the afferent arterioles. The ultrafiltrate accumulates in the Bowman’s space and thereafter traverses the tubules, where it is further modified into the final urine. During passage of tubular fluid down the renal tubule, solutes are reabsorbed by the highly selective transport mechanisms (Fig. 19.4). In general, most transport occurs in the proximal tubule, where the luminal membrane forms an elaborate ‘brush border’ of microvilli to provide extensive surface area for the reabsorptive processes. The brush borders are densely packed with mitochondria to supply the energy required for these active transports. Organic solutes such as low-molecular-weight proteins, sugars, and amino acids are avidly (>98%) reabsorbed in this segment. In addition, bulk transport of inorganic solutes and water is also accomplished in the proximal tubule. The subsequent tubules fine-tune the net reabsorption of these solutes and water. Dysfunction in the absorptive capacity of the tubules can give rise to various disorders of tubulopathy (see below). The glomerular filtration barrier consists of parietal epithelial cells with its foot process (podocytes), the glomerular basement membrane (GBM) and the capillary endothelial cells. Filtration of molecules across this structural barrier is limited by size, shape, and charge. Whereas charge selectivity is determined by negatively charged molecules present in the filtration barrier, size selectivity is determined by the GBM and by the slit diaphragm generated by interposing podocyte foot processes. The adult kidneys filter on average 150 litres of plasma per day containing 22.5 mol of sodium; more than 99% of filtered sodium is reabsorbed by the tubules, so that the final excretion is <1% (see Fig. 19.4). Disorders in sodium handling affect blood pressure (BP); sodium-losing disorders lead to hypotension and sodium-retaining disorders to hypertension. The Na+/K+-ATPase, which is present in all cells, is the driving force which generates a favourable electrochemical gradient for Na entry into the cell. This gradient also enables cotransport of other substances (such as glucose, amino acids, phosphate) into the cell. As sodium is the main determinant of intravascular volume, fractional excretion of sodium is normal in almost all renal salt-wasting disorders due to activation of renin–angiotensin–aldosterone system (RAAS). An overview of the different aetiologies of tubular disorder is given in Table 19.1. Table 19.1 Causes of renal tubular disorders Glucose transport Phosphate transport Amino acid transport Renal glycosuria Hypophosphataemic rickets Isolated, generalized aminoaciduria Proton (H+) secretion Sodium chloride transport Distal renal tubular acidosis Gitelman syndrome Water transport Sodium, potassium transport Nephrogenic diabetes insipidus Pseudohypoaldosteronism Liddle syndrome This syndrome describes a generalized proximal tubular disorder with at least initially well-preserved glomerular function. There is a long list of potential causes and it is helpful to split these into congenital, acquired and renal causes. The cardinal clinical features are growth faltering, polyuria and rickets in association with a normal plasma anion gap, metabolic acidosis, hypophosphataemia, hypokalaemia and generalized aminoaciduria. Supportive therapy may be required, which includes salt, water and nutritional supplementation as well as bicarbonate, electrolyte and phosphate replacement. Congenital causes include a familial idiopathic form, cystinosis (see below), tyrosinaemia and galactosaemia. Fanconi syndrome may also be acquired following treatment with aminoglycosides, sodium valproate, 6-mercaptopurine or ifosfamide or following poisoning with agents like toluene or paraquat. It occasionally occurs after renal transplantation, in the recovery phase of acute tubular necrosis or following tubulointerstitial nephritis or in focal and segmental glomerulosclerosis, a histopathological type of nephrotic syndrome. Nephropathic cystinosis is the commonest cause of Fanconi syndrome in Europe and North America. It is a disorder of lysosomal cystine transport of autosomal recessive inheritance, resulting in excessive intracellular accumulation of free cystine in many organs including the kidney, eyes and thyroids. Management includes specific therapy with mercaptamine, which prevents accumulation of lysosomal cystine. These syndromes occur as a result of disturbances in the thick ascending limb of loop of Henle (Bartter) and distal convoluted tubule (Gitelman), respectively, and typically result in a hypokalaemic, hypochloraemic metabolic alkalosis with salt wasting. The main problem is tubular loss of sodium and chloride and secondarily excess loss of potassium in the distal tubule associated with hyperreninaemia and hyperaldosteronism. In this part of the nephron, sodium reabsorption is linked to chloride reabsorption through the furosemide-sensitive sodium–potassium–chloride channel (NKCC2) in the loop of Henle (disrupted in Bartter syndrome) or the structurally similar thiazide-sensitive sodium–chloride channel (NCCT) in the early distal convoluted tubule (disrupted in Gitelman syndrome), respectively. Salt-wasting disorders from these parts of the nephron will always be associated with urinary chloride loss in excess of urine sodium loss because all chloride reabsorption is linked with sodium, with twice as much chloride as sodium reabsorbed via NKCC2. Moreover, sodium reabsorption but not chloride reabsorption can occur partly via the paracellular route and there is no capacity for chloride reabsorption more distally within the nephron. Hypercalciuria occurs in Bartter syndrome because calcium reabsorption is a linked paracellular process. Hypomagnesaemia does not occur because of compensatory reabsorption in the early distal convoluted tubule (DCT). By contrast, Gitelman syndrome has hypocalciuria and hypomagnesaemia because of a compensatory mechanism in the early DCT which down-regulates cells expressing NCCT (and an apical magnesium channel) in favour of cells which reabsorb sodium and calcium. Gitelman syndrome generally presents in older children or even adults with muscle weakness and cramps, and short stature. It is not uncommonly diagnosed following investigation of growth, constipation or enuresis. Classical Bartter syndrome is generally a more severe disorder, presenting in early childhood with growth faltering, dehydration, hypotonia and lethargy. There is often a history of maternal polyhydramnios with the classical form. The kidney achieves acid–base balance through bicarbonate reabsorption and acid secretion. Kidneys normally reabsorb up to 90% of filtered bicarbonate in the proximal tubules. The distal collecting tubules are principally responsible for acid secretion. Buffers in the tubular lumen bind free hydrogen ions, allowing excretion of the daily acid load within limits of the minimal achievable urine pH of 4.5 to 5. Ammonia and, to a lesser extent, phosphate are the main urinary buffers. Ammonia (NH3), which is formed from amino acid metabolism, can freely diffuse across tubular membranes, where it combines with protons to form ammonium (NH4+), which becomes trapped in the tubular lumen. Renal tubular acidosis (RTA) occurs in several ways: bicarbonate wasting in the proximal tubule (historically known as type 2 RTA), which almost always occurs as part of Fanconi syndrome; impairment in formation of ammonia results in type 4 RTA and in renal failure, where the acidosis is associated with hyperkalaemia; and failure to adequately secrete hydrogen ions is the primary defect in distal RTA (associated with hypokalaemia). In childhood distal RTA, most cases are genetic. Autosomal recessive forms can be associated with (ATP6V1B1) or without (ATP6V0A4) sensorineural deafness. Both mutations code for subunits of the H-ATPase apical hydrogen ion transporter. An autosomal dominant form is caused by mutations of the SLC4A1 gene which encodes the chloride–bicarbonate exchanger on the basolateral membrane. Urine pH in distal RTA is always >5.5, in contrast to proximal RTA where it varies according to the plasma bicarbonate. Initial correction of acidosis needs to take into account potassium and calcium, which will both decrease in response to alkali treatment. Maintenance treatment consists of sodium bicarbonate or citrate (sodium and/or potassium). Generally the doses of base required are less than for proximal acidosis. Children require lifelong follow-up and are at risk of nephrolithiasis and long-term deterioration in renal function from the nephrocalcinosis. In higher forms of vertebrates, including man, kidneys evolve through three stages: pronephros, mesonephros and metanephros. Pronephric and mesonephric kidneys are temporary and metanephros persist as permanent kidneys. Whereas the urinary tract develops from the cloaca and the intermediate mesoderm, the definitive functional kidney develops from the metanephros through involution of pronephros and mesonephros. Table 19.2 Congenital anomalies of the kidney and urinary tract Hydronephrosis is dilatation of the collecting system of the kidney, and hydroureter is dilatation of the ureter. Neither term implies obstruction. There is no clear definition or test for obstruction; its diagnosis requires a mixture of clinical and functional assessment including nuclear medicine imaging with DTPA/MAG3 scans (see section on imaging below). Hydronephrosis secondary to pelvi-ureteric junction obstruction (PUJO) is the commonest abnormality of the upper urinary tract. Renal ultrasound would show a dilated renal pelvis (isolated renal pelvis diameter >20 mm is highly suggestive of PUJO) with usually non-dilated ureter. It is more common in boys with overall incidence of 1 in 1000. Older children may present with acute loin or abdominal pain (Dietl’s crisis), haematuria, a palpable flank mass, infection (including pyonephrosis), nausea/vomiting, or pelvic rupture following minor trauma. As there can be progressive renal damage, early diagnosis and assessment of the degree of renal dysfunction is important. Dynamic renogram such as DTPA or MAG3 will aid in this as they give split renal function which can be monitored. As only around 25% develop clinical problems, the need for surgical intervention has to be carefully evaluated. Surgery is considered if there is deterioration of renal function or increasing dilatation of the renal pelvis with thinning of renal cortex. In fetal life, excretion is performed primarily by the placenta, which receives 50% of fetal cardiac output whereas the fetal kidney accounts for only 2–4%. Hence, creatinine at birth reflects maternal creatinine level. The glomerular filtration rate (GFR) is 10–15 mL/min/1.73m2 in the premature infant and 15–20 mL/min/1.73m2 in the term infant. These values double over the first two weeks after birth and reach adult values of 80–120 mL/min/1.73m2 by one year of age. The increase in GFR is achieved by recruitment of more glomeruli and not by new glomerulus generation. Urine formation starts by the end of the first trimester. Fetal urinary concentrating activity is poor and fetal urinary sodium and phosphate levels decrease while the creatinine increases with increasing gestation, reflecting increasing maturation. The maximum urine concentrating capacity is low even at birth (up to 600 mOsm/kg) which explains the higher susceptibility of newborns and infants to dehydration. Adult capacity is achieved only by 1–2 years. In contrast, the diluting capacity of newborns is equivalent to that of adults and the urine osmolality can be lowered to 30–50 mOsm/kg. For common developmental anomalies of the kidneys, see Box 19.1 and Figure 19.6. Urine examination is an important but simple and non-invasive aid to diagnosis. It should ideally start with visual inspection, which may provide clues to haematuria (red urine) or pyuria (cloudy urine). However, as other conditions can mimic them, suspected haematuria or pyuria on inspection needs to be confirmed by urine microscopy. Testing urine by dipstick is a useful bedside test. It can be used for: Glomerular filtration rate (GFR) is a measure of renal function. Although inulin remains the reference solute for GFR estimation, it is mainly used as a research tool. In clinical practice, GFR is measured using radiopharmaceuticals, such as 99mTc-DTPA, 125I-iothalamate and 51Cr-EDTA. Isotopic methods have the advantage that they do not require timed urine collection. In older children and adults, 24-hour urine collections are possible and can also be used. As these methods are too complex for routine clinical use, serum creatinine is used as a surrogate marker for renal function. However, this is not ideal as it is not an early indicator of renal damage. It also varies with age, muscle mass and nutritional status. Therefore, it can be inappropriately low in malnourished children or those with major amputation even in a stage of advanced renal failure. Formulas for calculating GFR (estimated GFR) have been devised to correct for body size, such as the modified Schwartz formula: where K is a constant (varying between 33 and 40). When testing for renal function, serum electrolytes are usually measured. They not only tell us about life-threatening complications (e.g. hyperkalaemia), but are useful in assessing the tubular function and volume status. For interpreting urinary loss of any electrolytes, a calculation of the fractional excretion is better than spot urinary value. Ultrasound scan (USS) is the most frequently used imaging modality in paediatric nephrology, as it is non-invasive and provides very good anatomical detail. Colour Doppler USS is useful for ascertaining renal blood flow, such as graft perfusion after renal transplantation, suspected renal vein or venous thrombosis or renal artery stenosis. Micturating cystourethrogram (MCUG) requires bladder catheterization. Once the catheter is in place, a small amount of contrast medium is injected through the catheter to fill up the bladder. It allows very good images to be obtained of the anatomy of the lower urinary tract and is the ‘gold standard’ test for documenting and grading vesico-ureteric reflux (see below). It is also useful in documenting any bladder outlet obstruction, such as posterior urethral valves. Its disadvantages are the small risk of infection, the requirement for radiation and the need for urethral catheterization that can be psychologically traumatic, particularly in the toddler age group. Dynamic scanning (MAG3 or DTPA scans) is useful for showing obstruction, such as pelvi-ureteric junction (PUJ) obstruction and is most often used after USS has shown dilated collecting system (hydronephrosis). It also gives an estimate of split differential function of both kidneys. MAG3 nuclear scan is supposed to be superior to 99mTc-DTPA scan and can even be used to document reflux in older children who are continent and can control urination on demand. Static scanning provides information about the renal parenchyma, most frequently used to identify renal scars. However, the radiation dose is higher than that in dynamic scanning. Fluid within the body is distributed between the intracellular fluid (ICF) and extracellular fluid (ECF) compartments (Fig. 19.7). Solute composition differs between the two components and this is maintained by cell membrane pump activity, and solute size and electrical charge.
Nephrology
Introduction
Anatomy and physiology of the kidney
Tubular disorders
Segment
Function
Disorder
Proximal tubule*
Ascending limb of Henle
Sodium, potassium, chloride transport
Bartter syndrome
Distal tubule
Collecting duct
Fanconi syndrome
Bartter and Gitelman syndromes
Renal tubular acidosis
Embryology of the urinary tract (Fig. 19.5)
Transient hydronephrosis (normal postnatal scan)
50%
Hydronephrosis with no evidence of obstruction or external pelvis
15%
Pelvi-ureteric junction obstruction (PUJO)
11%
Vesico-ureteric reflux (VUR)
9%
Megaureter (obstructed, refluxing, non-refluxing and non-obstructed or both refluxing and obstructed)
4%
Renal dysplasia
3%
Multicystic dysplastic kidney (MCDK)
2%
Duplex kidney ± ureterocoele
2%
Posterior urethral valves
1%
Others
5%
Hydronephrosis
Functional development
Investigating renal function
Urine examination
Glomerular filtration rate

Imaging of the urinary tract
Ultrasound
Micturating cystourethrogram
Nuclear medicine
Dynamic scanning (99mTc-DTPA/MAG3)
Static scanning (99mTc-DMSA)
Fluid and electrolyte homeostasis
Basic principles
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Nephrology
Chapter 19
Learning objectives
By the end of this chapter the reader should: