Neonatal Endocrinology
Elizabeth A. Catlin
Mary M. Lee
The transition from an intrauterine to an extrauterine environment at birth requires neonates to make rapid and appropriate homeostatic adaptations for independent living. The hormonal and metabolic responses of infants to stress—be it that of birth, surgery, or infection—at times may be more vigorous than those of adults. These overly robust endocrine–metabolic responses, and the neonate’s limited ability to modulate these responses, may be counterproductive in some situations. Alternatively, the neonate may respond inadequately with the rapid development of marked deviations of otherwise carefully protected body constituents. In either case, the clinical manifestations characteristic of older children or adults may be absent, making it difficult to appreciate a perturbation in endocrine function. This chapter covers common endocrine conditions and some of the more unusual endocrine disorders of newborns.
GLUCOSE BALANCE
The transfer of glucose from the placenta to the fetus is a concentration-dependent process driven by fetal glucose levels; this process is mediated by facilitative glucose transporter proteins. The fetus can generate some metabolic responses to nutritional states, although glucose is not produced, thus fetal growth and well-being depend on a maternal supply of glucose. At birth, neonatal plasma glucose values are initially 70% to 80% of maternal values. These values decline briefly before stabilizing by 2 to 3 hours of age. In healthy, term babies receiving enteral feedings by 3 hours of life, plasma glucose concentrations reach their nadir between 1 and 2 hours of life, at mean values of 56 mg/dL (SD +/- 19) (Fig. 62.1).
Intermittent and unpredictable feeding in the early neonatal period stresses the carbohydrate homeostatic capacity of a newborn. Developmental immaturity of two of the three counter-regulatory mechanisms—ketogenesis and gluconeogenesis—contributes to the increased risk of hypoglycemia in neonates. Moreover, enteral feeding may be needed to induce the expression of some of the enzymes necessary for ketogenesis. The third mechanism, hepatic gluconeogenesis, often is compromised by peripartum or neonatal stress. In healthy term and preterm neonates, insulin levels are elevated when compared with those of older children and appear to be less closely linked to glucose concentrations. The hepatic suppression of glucose production by insulin during glucose infusion, for example, has been noted to be delayed in newborns relative to that of adults. Extremely low-birth-weight infants (ELBW; infants of less than 1,000 g) have depressed processing of proinsulin as well as relative insulin resistance during hyperglycemia. Both relative insulin resistance and defective islet beta-cell processing of proinsulin are responsible for transient hyperglycemia in extremely preterm infants. Therefore, insulin’s role in neonatal glucoregulation may differ from its role in later development.
Hypoglycemia
Neonatal hypoglycemia is a common and typically transient problem in newborn infants that has traditionally been defined as a plasma glucose value of 40 mg/dL (2.2 mmol/L) or less. Controversy about the definition of neonatal hypoglycemia exists, however, and operational thresholds have been
proposed. We contend that, after the initial transient decline at birth, maintaining plasma glucose concentrations greater than 45 mg/dL during the first 24 hours and greater than 50 mg/dL thereafter is a sensible and conservative therapeutic goal, because fetal glucose levels are at least 40 mg/dL, and children and adults may have neuroglycopenic symptoms at glucose levels lower than this (as may neonates). Moreover, infants have an immature ketogenic capacity and are unable to generate ketones adequately as an alternative central nervous system fuel in response to hypoglycemia. Certain infants, including infants of diabetic mothers (IDMs), premature or growth-restricted infants, and perinatally stressed neonates especially are prone to the development of transient neonatal hypoglycemia. Persistent neonatal hypoglycemia is less common and is caused by congenital hyperinsulinism, deficits in counterregulatory hormones, or metabolic disorders that affect fasting adaptation.
proposed. We contend that, after the initial transient decline at birth, maintaining plasma glucose concentrations greater than 45 mg/dL during the first 24 hours and greater than 50 mg/dL thereafter is a sensible and conservative therapeutic goal, because fetal glucose levels are at least 40 mg/dL, and children and adults may have neuroglycopenic symptoms at glucose levels lower than this (as may neonates). Moreover, infants have an immature ketogenic capacity and are unable to generate ketones adequately as an alternative central nervous system fuel in response to hypoglycemia. Certain infants, including infants of diabetic mothers (IDMs), premature or growth-restricted infants, and perinatally stressed neonates especially are prone to the development of transient neonatal hypoglycemia. Persistent neonatal hypoglycemia is less common and is caused by congenital hyperinsulinism, deficits in counterregulatory hormones, or metabolic disorders that affect fasting adaptation.
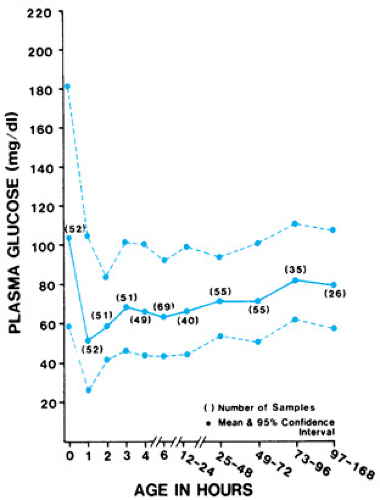 FIGURE 62.1. Predicted plasma glucose values during the first week of life in healthy term neonates with weights appropriate for their gestational age. (Reprinted with permission from Srinivasan G, Pildes RS, Cattamanchi G, et al. Plasma glucose values in normal neonates: a new look. J Pediatr 1986;109:114.) |
Pathophysiology
Persistence of the hyperinsulinemic state after birth in many IDMs who develop elevated insulin secretion in utero in response to maternal hyperglycemia causes self-limited hypoglycemia that can be severe but relatively asymptomatic (see Chapter 63, Infant of the Diabetic Mother). Infants with intrauterine growth restriction (IUGR) are at risk for neonatal hypoglycemia; 65% of premature and 25% of full-term IUGR babies become hypoglycemic. Factors contributing to their low plasma glucose levels include decreased glycogen and fat stores, large brain–to–body weight ratio, failure to mobilize alternative fuels adequately, and in some instances, a relative hyperinsulinemia. Premature neonates (less than 37 weeks’ gestation) also are at risk for the development of hypoglycemia. They have multiple contributing factors, such as lower total body energy stores, impaired ability to mobilize alternative fuels due to functional immaturity of the metabolic enzymes, and inadequate dietary intake. They also have an increased likelihood of having neonatal distress with sepsis, hypoxia, or perinatal depression, that can further compromise glucose homeostasis. ELBW babies must rely on gluconeogenesis and exogenous glucose infusion for glucose balance, because they have minimal glycogen available for glycogenolysis.
Hypoglycemia may develop in neonates with polycythemia-hyperviscosity with hematocrits greater than 65%. The greater red cell mass has increased glucose utilization, whereas less glucose is carried in a given volume of blood as a result of the diminished plasma fraction. The increased viscosity leads to sludging, which may leave tissues poorly oxygenated and, thus, more dependent on glucose for anaerobic metabolism. The hypoglycemia in these instances resolves with partial-exchange transfusion to reduce the hematocrit. Severe perinatal stress also predisposes to hypoglycemia; contributing features include excessive glucose utilization and exhaustion of stored substrates. Certain maternal therapies that suppress counter-regulatory mechanisms, such as treatment with propranolol, may contribute to inducing a hypoglycemic state in newborns.
Although rare, congenital hyperinsulinism is the most common cause of persistent hypoglycemia in infancy. Multiple genetic mutations have been identified thus far. Autosomal recessive mutations of the sulfonylurea receptors (SUR1) or associated potassium channels (Kir6.2) of pancreatic beta cells constitute the most severe forms of congenital hyperinsulinism with diffuse beta-cell disease. Mutations of either of these two adjacent genes on chromosome 11 affect the subunits of the KATP channel such that insulin secretion is constitutively active and unresponsive to ambient glucose. A sporadic form, due to loss of heterozygosity for the maternal allele and inheritance of an abnormal paternal allele for SUR1, causes focal beta-cell hyperplasia and is clinically indistinguishable from diffuse disease. These mutations of the ATP-dependent potassium channel are the most common cause of severe neonatal hyperinsulinism with the classic clinical picture of macrosomia, increased insulin/glucose ratios, and a requirement for high glucose infusion rates. Autosomal dominant hyperinsulinism appears to be a milder disease. Although the majority of cases have not been associated with particular mutations, a gain-of-function mutation of the glucokinase gene can cause a mild form of congenital hyperinsulinism that is responsive to diazoxide. Congenital hyperinsulinism with associated hyperammonemia is caused by an activating mutation in the glutamate dehydrogenase gene. These infants typically present later in infancy and have leucine-sensitive hypoglycemia with mild fasting defects.
Neonates with the Beckwith-Wiedemann syndrome (Fig. 62.2) have macroglossia, characteristic ear creases or pits, visceromegaly, omphalocele, and are large for gestational age (LGA). Nearly 50% of reported cases exhibit hyperinsulinemic hypoglycemia caused by diffuse islet cell hyperplasia. Hypoglycemia in such infants may be severe and long-lasting, necessitating pharmacologic management of their hyperinsulinism in addition to glucose supplementation.
Hypopituitarism and isolated growth hormone (GH) deficiency are other endocrine causes of persistent hypoglycemia in infancy. Although growth may remain normal during infancy, associated physical features, such as midline defects or microphallus, are helpful diagnostically. Very infrequently, neonatal hypoglycemia is caused by one of the inborn errors of metabolism in the gluconeogenic or glycogenolysis pathways, such as galactosemia or type I glycogen storage disease (see Chapter 387, Disorders of Carbohydrate Metabolism). Genetic defects of the glucose transporter 1, which transfers glucose across the blood–brain barrier, have been recognized as a cause of central nervous system hypoglycemia and seizures despite normal blood glucose levels.
Diagnosis
Clinicians must have a low threshold for suspecting hypoglycemia in neonates, because the symptoms are subtle.
Affected infants may be jittery or manifest such nonspecific symptoms as apnea, seizures, poor feeding, or lethargy, but they may be also essentially asymptomatic. The neurocognitive sequelae of asymptomatic neonatal hypoglycemia are unknown. A careful pregnancy history and physical examination may reveal evidence of growth restriction, visceromegaly, or other clues to the etiology of the hypoglycemia. A plasma glucose level should be obtained; if capillary sampling is used, care must be taken to warm the heel adequately. If the glucose concentration is 40 mg/dL or less in asymptomatic infants or less than 45 mg/dL in infants with symptoms, assessment and possibly treatment should be instituted. Initial screening can be carried out using glucose oxidase strips (e.g., Dextrostix, Chemstrips) or with a bedside glucose analyzer, but laboratory confirmation of low values is required. The strips should be fresh or packaged individually, because strips from open bottles may be inaccurate and give erroneously low values.
Affected infants may be jittery or manifest such nonspecific symptoms as apnea, seizures, poor feeding, or lethargy, but they may be also essentially asymptomatic. The neurocognitive sequelae of asymptomatic neonatal hypoglycemia are unknown. A careful pregnancy history and physical examination may reveal evidence of growth restriction, visceromegaly, or other clues to the etiology of the hypoglycemia. A plasma glucose level should be obtained; if capillary sampling is used, care must be taken to warm the heel adequately. If the glucose concentration is 40 mg/dL or less in asymptomatic infants or less than 45 mg/dL in infants with symptoms, assessment and possibly treatment should be instituted. Initial screening can be carried out using glucose oxidase strips (e.g., Dextrostix, Chemstrips) or with a bedside glucose analyzer, but laboratory confirmation of low values is required. The strips should be fresh or packaged individually, because strips from open bottles may be inaccurate and give erroneously low values.
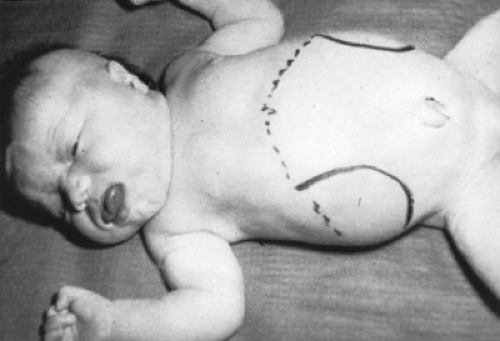 FIGURE 62.2. Neonate with Beckwith-Wiedemann syndrome. This 38 weeks’ gestation, 34-cm, 5.218-kg plethoric infant was the third child in this family to be affected with the syndrome. Note, in addition to his large size and abundance of subcutaneous fat, the enlarged tongue and umbilical hernia. The enormously enlarged, multilobular kidneys have been outlined with a skin pencil. The vertical creases on the lobuli of the ears are not shown well. The infant required prolonged infusion of intravenous glucose to prevent hyperinsulinemic hypoglycemia and required a partial glossectomy and repair of the umbilical hernia. |
Therapy
Stable and relatively mature babies may be treated safely with early enteral feedings and careful monitoring of subsequent plasma glucose values. If blood glucose is greater than 45 mg/dL, normal feeding may be started as soon as the infant’s condition permits, with capillary glucose monitoring every 1 to 2 hours until blood sugars stabilize in the normal range. If the glucose value is between 25 and 45 mg/dL, a repeat sample is obtained while the baby is given 10 to 15 mL of formula (orally or by gavage feeding) with frequent monitoring of the blood glucose until it is stable (greater than 40 to 45 mg/dL). If the blood glucose is less than 25 mg/dL, parenteral glucose supplementation is required, first administering a “mini-bolus” of 10% glucose, 2 to 3 mL/kg intravenously, followed by a constant infusion of 10% glucose delivered at a rate of 4 to 6 mg/kg/minute, which approximates the basal glucose production rate of the healthy neonate. Glucose values must be monitored closely, with titration of the infusate to maintain normoglycemia.
Infants with transient hypoglycemia or hypoglycemia attributable to one of the self-limited disorders described require supportive therapy to maintain glucose levels in the normal range, but minimal diagnostic evaluation is necessary. Infants with prolonged hypoglycemia, those requiring high glucose infusions (more than 10 to 15 mg/kg/minute), and those with a clinical course suspicious for a pathologic disorder of glucose regulation need further workup. Inappropriately high serum insulin levels (relative to simultaneously obtained plasma glucose levels) without evidence of Beckwith-Wiedemann syndrome or maternal diabetes mellitus suggest hyperinsulinism. Insulin secretion should be suppressed completely during a hypoglycemic episode; thus, the finding of measurable concentrations of insulin and absent ketone production is suspicious for hyperinsulinism. A glucagon stimulation test can help identify a hyperinsulinemic state. In a hypoglycemic infant, the administration of 0.5 mg of glucagon will not raise significantly the plasma glucose, unless glycogen breakdown is being suppressed by insulin (hyperinsulinism) or decreased counter-regulatory hormones (panhypopituitarism). An increase in glucose of more than 30 mg/dL is indicative of glycogen reserve. Therefore, obtaining a blood sample for ketones, free fatty acids, cortisol, and GH, in addition to the insulin level, during hypoglycemia is essential. Evaluation of the gluconeogenic or glycogenolytic pathway may be necessary in rare cases suspicious for these disorders. Fasting studies are at times necessary to assess fasting tolerance and to make a definitive diagnosis.
Infants with hyperinsulinemic hypoglycemia and infants with IUGR may require especially high glucose infusions (15 mg/kg/minute or higher), which usually are delivered via a central venous catheter. Intravenous therapy should be maintained until the glucose level has stabilized. Rebound hypoglycemia in IDMs can be avoided by a slow tapering of the intravenous glucose being administered and the maintenance of the blood glucose in the normal range, avoiding both hypoglycemia and hyperglycemia. If intravenous access is difficult in IDMs, glucagon (0.3 mg/kg to a maximum dose of 1 mg) can be given to override the inhibitory effect of insulin on glycogenolysis and raise an affected infant’s blood glucose level within 10 to 15 minutes of the injection. It must be emphasized that glucagon benefits only infants with a large glucose reservoir in the form of glycogen; it is ineffective in low-birth-weight or perinatally depressed infants who have depleted their glycogen stores.
Infants who require very high glucose infusions may need pharmacologic therapy to aid in the treatment of hypoglycemia. Diazoxide suppresses insulin secretion and is the initial choice for hyperinsulinism; the usual dosage is 10 to 15 mg/kg/day. Patients with SUR1 or Kir6.2 mutations, however, are unlikely to respond adequately to diazoxide and generally require a subtotal 95% to 99% pancreatectomy. Octreotide, a somatostatin analogue, suppresses insulin release and has been useful in conjunction with diazoxide in certain patients for the long-term management of hyperinsulinemia. Glucagon infusions (5 to10 μg/kg/hour) can be helpful as a temporizing measure, particularly preoperatively, but the action of these infusions are not sustained for chronic use. At some centers, focal lesions can be localized intraoperatively and successfully resected. Postoperatively, pharmacologic intervention still may be necessary for optimal glucose control.
Hyperglycemia
Hyperglycemia in neonates is defined as random plasma glucose concentrations greater than 150 mg/dL. The incidence of hyperglycemia has increased in parallel with the increased survival of very premature and high-risk infants. Some studies suggest that hyperglycemia contributes to increased morbidity and mortality in neonates.
Pathophysiology
ELBW infants have a relative insulin insensitivity and delayed processing of proinsulin during hyperglycemia. Other conditions associated with transient hyperglycemia in the newborn period include sepsis, surgical stress, hypoxia, central nervous system insults (including intracranial hemorrhage), and treatment with methylxanthines. Stress-related hyperglycemia results, in part, from elevated catecholamine and cortisol levels. Pancreatic agenesis is a rare cause of neonatal glucose intolerance and is caused by insulin promoter factor 1 mutations; these babies also exhibit significant IUGR. Neonatal diabetes, estimated to occur once in 500,000 births, has a highly variable course. Affected babies may have transient diabetes, with or without periods of remission, or permanent diabetes, and most are small for gestational age. A complete deficiency of glucokinase has been found to cause permanent neonatal diabetes. The small size of these neonates, in contrast to the macrosomia of IDMs, is believed to reflect insulin’s important role as an intrauterine growth hormone.
Therapy
In a hyperglycemic neonate (greater than 150 mg/dL), the first measure is to calculate the glucose infusion rate in mg/kg/minute and gradually decrease the rate by 1 to 2 mg/kg/minute every 3 to 4 hours, monitoring glucose levels 30 to 60 minutes after changing the rate. Hypotonic infusates must be avoided. Insulin therapy should be considered if hyperglycemia persists despite the administration of glucose at basal production rates (approximately 4 to 6 mg/kg/minute). Fluid
balance, glycosuria, blood pressure, oxygenation, and perfusion should be checked carefully and, if sepsis is suspected, appropriate cultures should be obtained and antibiotics given. In very low-birth-weight infants (of less than 1.5 kg), consider measures to minimize insensible fluid losses, such as transfer from radiant warmers to isolettes. If hyperglycemia persists (greater than 200 mg/dL), therapy may be required using a continuous infusion of regular insulin, starting at 0.01 to 0.02 units/kg/hour, with the aim of maintaining plasma glucose at 100 to 150 mg/dL and taking care to avoid hypoglycemia. ELBW infants often require insulin infused continuously, along with intravenous glucose containing hyperalimentation, to permit the delivery of adequate nutritional substrates for growth. A larger and persistent requirement for insulin generally distinguishes the rare neonate with diabetes mellitus from the infant mounting a vigorous stress response who has transient hyperglycemia that resolves within a few days.
balance, glycosuria, blood pressure, oxygenation, and perfusion should be checked carefully and, if sepsis is suspected, appropriate cultures should be obtained and antibiotics given. In very low-birth-weight infants (of less than 1.5 kg), consider measures to minimize insensible fluid losses, such as transfer from radiant warmers to isolettes. If hyperglycemia persists (greater than 200 mg/dL), therapy may be required using a continuous infusion of regular insulin, starting at 0.01 to 0.02 units/kg/hour, with the aim of maintaining plasma glucose at 100 to 150 mg/dL and taking care to avoid hypoglycemia. ELBW infants often require insulin infused continuously, along with intravenous glucose containing hyperalimentation, to permit the delivery of adequate nutritional substrates for growth. A larger and persistent requirement for insulin generally distinguishes the rare neonate with diabetes mellitus from the infant mounting a vigorous stress response who has transient hyperglycemia that resolves within a few days.
CALCIUM BALANCE
The placenta actively transports calcium to the fetus against a concentration gradient under the regulation of parathyroid hormone–related peptide. The majority of fetal skeletal mineralization takes place during the last trimester. Serum calcium in the newborn, under normal circumstances, decreases for several days after birth, stabilizes, and gradually increases to the concentrations found in older infants and children.
Hypocalcemia
Hypocalcemia occurs commonly in the neonatal period but is generally transient and rarely indicative of a persistent metabolic disorder. Neonatal hypocalcemia is generally subdivided into early and late-onset categories (see Chapter 64, Mineral Metabolism in the Newborn). Early hypocalcemia has been defined as a total serum calcium concentration of less than 8 mg/dL (2.0 mmol/L) in the full-term neonate or less than 7 mg/dL (1.75 mmol/L) in the premature neonate, or, perhaps more precisely, an ionized calcium level of less than 1.0 mmol/L (4 mg/dL) during the first few days of life. Inasmuch as 50% of calcium in serum is bound loosely to protein and varies according to the serum protein concentration, pH, and other factors, the biologically active ionized calcium concentration is a better indicator of the calcium status. The postnatal serum calcium nadir at 24 and 48 hours of life normally stimulates the secretion of parathyroid hormone (PTH). The inadequate PTH response of premature neonates, IDMs, and infants with perinatal depression to the decreasing calcium concentrations underlies their susceptibility to early hypocalcemia.
Pathophysiology
Late neonatal hypocalcemia occurs toward the end or after the first week of life and may be caused by congenital hypoparathyroidism, hypomagnesemia, and high-phosphate formula consumption (synonymous with late infantile tetany). Congenital hypoparathyroidism is caused usually by agenesis or dysgenesis of the parathyroid glands, and often is associated with the DiGeorge syndrome or chromosome 22q11 microdeletion syndrome. A transient hypoparathyroidism may be present in infants of women with hyperparathyroidism. Rarely, intrauterine nutritional vitamin D deficiency from severe maternal vitamin D deficiency or hereditary vitamin D–resistant rickets can present with late-onset hypocalcemia. Late-onset hypocalcemia associated with hypomagnesemia may occur in small-for-gestational-age neonates, in those with hepatic disease or small-bowel resections and, very rarely, in infants with magnesium malabsorption or disorders of renal magnesium handling. Hypomagnesemia causes late hypocalcemia via several mechanisms: It inhibits parathyroid hormone (PTH) secretion, induces a relative end-organ insensitivity to PTH, and causes decreased calcium absorption and decreased exchange of magnesium for calcium at the bone surface.
Diagnosis
The diagnosis of neonatal hypocalcemia begins with an assessment of history, clinical signs, and laboratory data. The majority of babies with early neonatal hypocalcemia are asymptomatic and require no treatment except for close follow-up and monitoring. Hypocalcemic neonates may occasionally exhibit seizure activity, tremors, tetany or lethargy, and poor feeding. Monitoring of serum calcium, phosphorus, and magnesium with a careful assessment of intake is indicated in high-risk infants. Late hypocalcemia is more likely to be associated with a chronic condition and, in addition to the laboratory tests mentioned, PTH and vitamin D levels may be helpful diagnostically.
Therapy
Although many neonatal units treat at-risk neonates with early calcium supplementation, the indications for this practice are not clear. Symptomatic early hypocalcemia is usually transient and may require temporary therapy with calcium supplementation. Oral and intravenous calcium supplementation for early hypocalcemia can be achieved with elemental calcium, 75 mg/kg/day as an initial oral dose or 24 to 75 mg/kg/day parenterally, usually given for less than 3 days. The subacute and chronic conditions of late hypocalcemia are treated with calcitriol (1,25-dihydroxyvitamin D), starting with a dose of 0.125 μg once or twice daily and calcium supplementation as described (see Chapter 64). Parathyroid agenesis requires lifelong therapy to prevent hypocalcemia. During the initiation of therapy, serum calcium and phosphorus should be monitored daily to maintain their concentrations in the low-normal range.
Seizures associated with hypocalcemia are treated with intravenous 10% calcium gluconate, 1 to 2 mL/kg, given slowly over 5 to 10 minutes to avoid inducing bradycardia. Care must be taken with the intravenous dosing of calcium-containing solutions to avoid extravasation and associated tissue damage.
Hypercalcemia
Pathophysiology
Neonatal hypercalcemia usually is the result of excessive calcium supplementation, especially in very premature newborns. Babies with Williams syndrome (idiopathic infantile hypercalcemia syndrome with elfin facies and supravalvular aortic stenosis) may have hypercalcemia and nephrocalcinosis, which typically resolves spontaneously by 4 years of age. Primary hyperparathyroidism, although extremely rare, is caused by homozygous mutations of the plasma membrane calcium-sensing receptor (CASR). Heterozygotes for mutations of this receptor have familial hypocalciuric hypercalcemia, which is often asymptomatic, but homozygotes have a severe form of hypercalcemia, which is often lethal.
Therapy
The treatment of neonatal hypercalcemia should be prompt and includes hydration at one-and one-half to two times maintenance using 5% dextrose with 0.5 normal saline and potassium chloride (3 mEq/dL); furosemide given at 1 mg/kg per dose intravenously twice or three times daily to inhibit renal
tubular calcium resorption; and phosphate supplementation to maintain normal serum phosphorus values. Vitamin D and calcium intake should be limited, and hydrocortisone may help decrease the intestinal absorption of calcium. Primary hyperparathyroidism often requires surgical resection of the parathyroid glands for definitive management.
tubular calcium resorption; and phosphate supplementation to maintain normal serum phosphorus values. Vitamin D and calcium intake should be limited, and hydrocortisone may help decrease the intestinal absorption of calcium. Primary hyperparathyroidism often requires surgical resection of the parathyroid glands for definitive management.
MAGNESIUM BALANCE
Critically ill neonates and IDMs in poor glycemic control or substance-abusing mothers are at risk for hypomagnesemia. Maternal nutritional status may contribute to the depressed total serum magnesium levels measured in such neonates, and increased urinary magnesium losses are associated with the diabetic state. A potential problem in the interpretation of magnesium data is that of definition. Whereas magnesium exists in the extracellular compartment in ionized, protein-bound, and miscellaneously bound forms (similar to calcium), hypomagnesemia generally has been defined on the basis of total magnesium levels. Because the ionized fraction of magnesium is considered to be physiologically more relevant, determinations of total magnesium may not represent a valid parameter of neonatal magnesium balance.
Pathophysiology
Neonatal hypomagnesemia may result from inadequate nutritional supplementation in neonates treated with parenteral alimentation or magnesium malabsorption, either associated with surgical short bowel or caused by a rare X-linked defect presenting after the first few weeks of life and requiring lifelong magnesium replacement. Hypomagnesemia caused by renal wasting may be seen as a result of aminoglycoside therapy or as an inherited disorder of renal magnesium handling. IUGR and hepatic disease predispose to neonatal hypomagnesemia. Low magnesium concentrations reduce both the secretory response of the parathyroid gland to hypocalcemia and the end-organ responsiveness to PTH; thus, refractory hypocalcemia may be the first clue to a primary magnesium deficit.
Hypermagnesemia (total serum magnesium levels of greater than 2.1 mEq/L or 1.05 mmol/L) is typically iatrogenic. It can be seen in the babies of mothers being treated with magnesium sulfate or magnesium-containing antacids. Often, a functional ileus is present, and generalized central nervous system depression may result in lethargy, apnea, and diminished responsiveness. Alternatively, parenteral hyperalimentation may provide a relative excess of magnesium.
Diagnosis
Signs of neuromuscular irritability, which may be evident in neonates with hypomagnesemia, may aid in diagnosis. Studies of infants with seizures in the first week after birth revealed that as many as one-half of all neonates with hypocalcemic seizures had coexistent hypomagnesemia.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


