Neonatal Cholestasis
Donald A. Novak
Frederick J. Suchy
William F. Balistreri
Neonatal cholestasis, defined as prolonged conjugated hyperbilirubinemia, is the end result of impaired bile flow and excretion. The cumulative incidence of neonatal cholestasis is near 1 in 2,500 live births. Liver dysfunction in the neonate, regardless of the cause, commonly is associated with bile secretory failure and cholestatic jaundice. The potential mechanisms by which bile secretion may be impaired are many. Hepatocellular injury, such as that noted in neonatal hepatitis, may cause functional impairment of bile secretion. Mechanical obstruction to bile flow also may occur, as noted in biliary atresia. Although many disorders can present as neonatal cholestasis, neonatal hepatitis and biliary atresia are the most common syndromes, together accounting for the majority of cases of prolonged conjugated hyperbilirubinemia in infants (Box 56.1).
BOX 56.1 Differential Diagnosis of Conjugated Hyperbilirubinemia (Neonatal Cholestasis)
Bile Duct Obstruction
Cholangiopathies
Biliary atresia
Choledochal cyst
Neonatal sclerosing cholangitis
Spontaneous perforation of common bile duct
Bile duct stenosis
Caroli disease
Other
Inspissated bile-mucus plug
Cholelithiasis
Tumors or masses (intrinsic and extrinsic)
Neonatal Hepatitis
“Idiopathic”
Viral hepatitis
Cytomegalovirus
Rubella
Herpesviruses
Simplex
Zoster
Human herpesvirus type 6
Adenovirus
Enteroviruses
Parvovirus B19
Hepatitis B
? Non-A, non-B, non-C hepatitis
Human immunodeficiency virus
Bacterial and parasitic hepatitis
Bacterial sepsis
Syphilis
Listeriosis
Tuberculosis
Toxoplasmosis
Cholestatic Syndromes
Alagille syndrome
Progressive familial intrahepatic cholestasis
Type 1: Familial intrahepatic cholestasis 1
Type 2: BSEP (ATP-dependent bile acid transporter)
Type 3: MDR3 deficiency
Hereditary cholestasis with lymphedema (Aagenaes syndrome)
Benign recurrent intrahepatic cholestasis: Familial intrahepatic cholestasis 1
Disorders of bile acid synthesis
Peroxisomal disorders
Zellweger syndrome
Familial cholestasis of North American Indians
Metabolic Diseases
Disorders of amino acid metabolism
Tyrosinemia
Disorders of lipid metabolism
Niemann-Pick disease type C
Gaucher disease
Wolman disease/cholesteryl ester storage disease
Disorders of the urea cycle
Arginase deficiency
Disorders of carbohydrate metabolism
Galactosemia
Fructosemia
Type IV glycogenosis
Carbohydrate deficient glycoprotein syndrome
Disorders of oxidative phosphorylation
Alpha-1-antitrypsin deficiency
Cystic fibrosis
Hypopituitarism (septooptic dysplasia)
Hypothyroidism
Neonatal iron storage disease
Toxic Conditions
Drugs
Parenteral nutrition
Miscellaneous Associations
Shock, hypoperfusion
Langerhans cell histiocytosis
Intestinal obstruction
Erythrophagocytic lymphohistiocytosis
Neonatal lupus erythematosus
Indian childhood cirrhosis
Extracorporeal membrane oxygenation
Autosomal trisomies
Graft-versus-host disease
Venoocclusive disease
Identification of the infant with cholestasis begins with measurement of total and conjugated bilirubin fractions (Fig. 56.1). The cholestatic infant will have an elevated conjugated fraction, generally at a level of more than 2 mg/dL and accounting for more than 15% to 20% of the total bilirubin. The possibility of liver or biliary tract disease must be considered in any neonate jaundiced beyond 2 weeks of age. Indeed, jaundice after 11 days in term infants and after 14 days in premature infants is unusual, occurring in only 0.5% of infants in one study. Subsequent efforts must be directed toward rapid diagnosis of and therapy for potentially treatable disorders, such as sepsis, galactosemia, and hypothyroidism or panhypopituitarism, in which delay of diagnosis may have catastrophic consequences. Next, differentiation of biliary atresia from neonatal hepatitis must be accomplished, because the former requires surgical intervention. In all patients with cholestasis, effective management of the consequences of chronic cholestasis is imperative (Fig. 56.2). Early in the workup of any cholestatic infant, vitamin K (2.5 mg) should be given in an effort to prevent life-threatening hemorrhage. Further description of specific diagnostic modalities is included with the discussion of each entity.
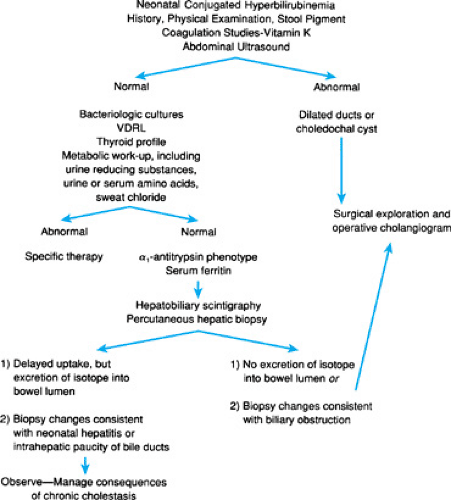 FIGURE 56.1. Flow chart for the workup of neonatal cholestasis. |
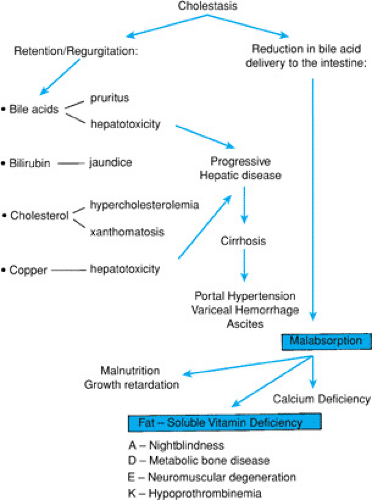 FIGURE 56.2. Consequences of chronic cholestasis. |
INFECTIOUS CAUSES OF CHOLESTASIS
That cholestasis may occur in patients with gram-negative bacterial infection has long been recognized. The incidence of jaundice during episodes of neonatal sepsis has been estimated at 20% to 60%. Urinary tract infections caused by Escherichia coli frequently have been associated with jaundice, particularly in male infants. The infants may appear clinically well and may have jaundice as their only symptom. Other bacteria, including gram-negative rods, staphylococci, streptococci, and Listeria, have been implicated in cholestasis. Limited autopsy studies have shown centrilobular hepatic necrosis and canalicular bile stasis; these features and direct experimental evidence suggest that bacterial toxins produce canalicular dysfunction, probably through inhibition of the canalicular multiple organic anion transporter. Jaundice is reversible with successful therapy for the underlying infection. Thus, cultures (blood and urine in particular) are important components of the initial evaluation phase for neonates with cholestasis.
Other unusual infectious causes of neonatal cholestasis include any of the so-called TORCH (toxoplasmosis, other viruses, rubella, cytomegalovirus [CMV], herpes) agents, as well as coxsackieviruses and echoviruses. Parvovirus B19 and human herpesvirus type 6 are associated with cholestatic liver disease. The infant with congenital toxoplasmosis may be noted at birth with hepatomegaly (60%) and hyperbilirubinemia (40%), or these features may develop later in the neonatal period. Although hepatic disease generally is mild, progressive hepatic dysfunction may occur. Hepatic pathologic features are nonspecific and include mononuclear periportal inflammation and canalicular bile stasis. Diagnosis may be made using utilizing serologic assays or through demonstration of the parasite in spinal fluid sediment. Antiparasitic therapy may arrest disease progression.
Congenital rubella may be associated with hepatomegaly in 65% of cases and with jaundice in 15% of cases. The clinical presentation and hepatic pathologic findings are nonspecific. Elevation of aminotransferase values may occur, as may acholic stools. The prognosis for the hepatic disease is good, with progression to hepatic fibrosis and failure uncommon. Diagnosis is through serology and viral isolation. No specific therapy is available.
CMV infection may produce symptoms in the neonate of hepatosplenomegaly and, occasionally, hyperbilirubinemia, in addition to the other well-known stigmata of congenital CMV. In the jaundiced infant with abnormal serum aminotransferase levels, CMV infection is suggested by liver biopsy findings of
focal areas of hepatocyte necrosis and portal inflammatory infiltrates composed of lymphocytes and neutrophils. Intranuclear viral inclusions may be noted in bile duct epithelial cells and (rarely) in hepatocytes. In addition, giant cell transformation, bile stasis, and extramedullary hematopoiesis may be noted. Diagnosis is through isolation of the organism from urine or tissue. Detection of infection through the use of polymerase chain reaction or monoclonal antibody staining in tissue sections is also available. The prognosis for CMV-related hepatic disease generally is good, with rare progression to severe chronic liver disease. Ganciclovir therapy may be indicated for severe congenital disease attributed to CMV.
focal areas of hepatocyte necrosis and portal inflammatory infiltrates composed of lymphocytes and neutrophils. Intranuclear viral inclusions may be noted in bile duct epithelial cells and (rarely) in hepatocytes. In addition, giant cell transformation, bile stasis, and extramedullary hematopoiesis may be noted. Diagnosis is through isolation of the organism from urine or tissue. Detection of infection through the use of polymerase chain reaction or monoclonal antibody staining in tissue sections is also available. The prognosis for CMV-related hepatic disease generally is good, with rare progression to severe chronic liver disease. Ganciclovir therapy may be indicated for severe congenital disease attributed to CMV.
Herpes simplex virus may cause jaundice and massive hepatic necrosis, with liver failure in conjunction with the other clinical features of neonatal herpetic infection. Coxsackievirus and echovirus infection (types 11, 14, 19, and 20) may present in similar fashion. Diagnosis in each case is through viral isolation, serology, and polymerase chain reaction. Therapy for documented neonatal herpes simplex infection consists of intravenous acyclovir.
Eighty percent of infants with congenital syphilis have hepatomegaly, whereas 40% are jaundiced. Hepatic biopsy may reveal extramedullary hematopoiesis, parenchymal or portal inflammatory infiltrates, and granulomatous lesions. Spirochetes may be evident; however, the diagnosis typically is made by serologic evaluation. Penicillin remains essential in the therapy of affected infants, but it may exacerbate hepatic disease resulting from syphilis.
INTRAHEPATIC CHOLESTASIS
Intrahepatic cholestasis is a term that encompasses a diverse group of disorders, some associated with “paucity” of bile ducts, some associated with disordered bile acid metabolism, and others with abnormalities of transport across the hepatocyte membrane. Still others remain undefined. These disorders may present in the neonatal period with jaundice or (later in life) with chronic cholestasis. The term progressive familial intrahepatic cholestasis (PFIC) is used to describe a heterogeneous group of autosomal recessive progressive liver disorders in which cholestasis of hepatocellular origin often presents in the neonatal period or the first year of life and leads to death from liver failure at ages ranging from infancy to adolescence. However, it has long been thought, because the clinical, biochemical, and pathologic features and natural progression are variable, that this group has significant heterogeneity. Recent molecular and genetic studies have allowed the identification of genes responsible for three types of PFIC and have shown that these phenotypes are related to mutations in hepatocellular transport systems involved in bile formation. Inherited or acquired dysfunction of the mechanisms involved in the generation of bile flow, especially of canalicular transport proteins, therefore results in substrate retention manifest as cholestasis. A proposed classification of the various disorders comprising intrahepatic cholestasis is depicted in Box 56.2.
Disorders of Canalicular Transport
PFIC 1, or Byler syndrome, is a progressive familial disorder in which jaundice, at first episodic, becomes constant by 1 to 4 years of age. Pathologic study of the liver reveals progressive cholestasis and cirrhosis. Affected children have steatorrhea, failure to thrive, and rickets. Laboratory studies reflect severe cholestasis; serum gamma-glutamyltranspeptidase levels are paradoxically normal. Death occurs in childhood and typically results from the consequences of hepatic failure and portal hypertension. Some patients with this disorder have benefited from partial cutaneous biliary diversion, which may promote the loss of potentially hepatotoxic bile acids. A gene associated with Byler disease and the original Amish kindred in whom this disorder was defined were mapped to chromosome 18q21 and subsequently shown to be the region encoding FIC1. FIC1 is a type IV P-type adenosine triphosphatase (ATPase), thought to function as an ATP-dependent amino phospholipid translocase. The protein is located on the canalicular membrane of hepatocytes, as well as in cholangiocytes. Also associated with mutations in FIC1 is benign recurrent intrahepatic cholestasis, with onset generally occurring during childhood. The clinical course of this disorder includes recurrent episodes of cholestasis that resolve spontaneously. The mechanism by which alterations in FIC1 cause the phenotype of benign recurrent intrahepatic cholestasis, or those associated with PFIC 1 (see earlier), remain unclear. Although FIC1 does not appear to be a bile acid transporter, mutations in FIC1 are associated with a profound reduction of biliary bile salts.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


