Chapter 601 Muscular Dystrophies
601.1 Duchenne and Becker Muscular Dystrophies
Clinical Manifestations
Infant boys are only rarely symptomatic at birth or in early infancy, although some are mildly hypotonic. Early gross motor skills, such as rolling over, sitting, and standing, are usually achieved at the appropriate ages or may be mildly delayed. Poor head control in infancy may be the first sign of weakness. Distinctive facies are not an early feature because facial muscle weakness is a late event; in later childhood, a “transverse” or horizontal smile may be seen. Walking is often accomplished at the normal age of about 12 mo, but hip girdle weakness may be seen in subtle form as early as the 2nd year. Toddlers might assume a lordotic posture when standing to compensate for gluteal weakness. An early Gowers sign is often evident by age 3 yr and is fully expressed by age 5 or 6 yr (see Fig. 584-5). A Trendelenburg gait, or hip waddle, appears at this time. Common presentations in toddlers include delayed walking, falling, toe walking and trouble running or walking upstairs, developmental delay, and, less often, malignant hyperthermia after anesthesia.
Diagnosis
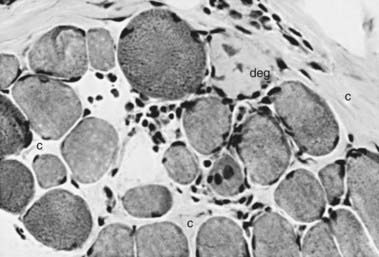
The muscle biopsy is diagnostic and shows characteristic changes (Figs. 601-1 and 601-2). Myopathic changes include endomysial connective tissue proliferation, scattered degenerating and regenerating myofibers, foci of mononuclear inflammatory cell infiltrates as a reaction to muscle fiber necrosis, mild architectural changes in still-functional muscle fibers, and many dense fibers. These hypercontracted fibers probably result from segmental necrosis at another level, allowing calcium to enter the site of breakdown of the sarcolemmal membrane and trigger a contraction of the whole length of the muscle fiber. Calcifications within myofibers are correlated with secondary β-dystroglycan deficiency.
Genetic Etiology and Pathogenesis
Despite the X-linked recessive inheritance in DMD, about 30% of cases are new mutations, and the mother is not a carrier. The female carrier state usually shows no muscle weakness or any clinical expression of the disease, but affected girls are occasionally encountered, usually having much milder weakness than boys. These symptomatic girls are explained by the Lyon hypothesis in which the normal X chromosome becomes inactivated and the one with the gene deletion is active (Chapter 75). The full clinical picture of DMD has occurred in several girls with Turner syndrome in whom the single X chromosome must have had the Xp21 gene deletion.
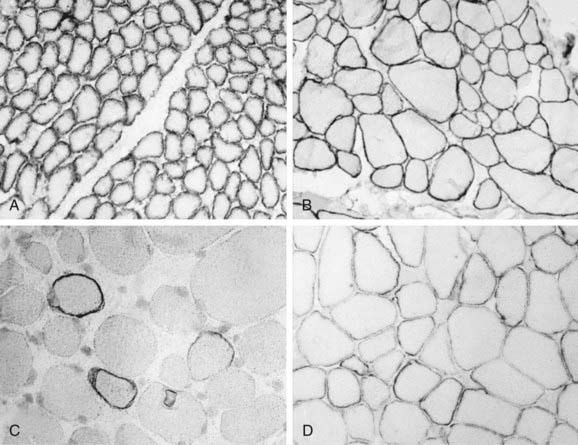
Analysis of the dystrophin protein requires a muscle biopsy and is demonstrated by Western blot analysis or in tissue sections by immunohistochemical methods using either fluorescence or light microscopy of antidystrophin antisera (see Fig. 601-2). In classic DMD, levels of <3% of normal are found; in BMD, the molecular weight of dystrophin is reduced to 20-90% of normal in 80% of patients, but in 15% of patients the dystrophin is of normal size but reduced in quantity, and 5% of patients have an abnormally large protein caused by excessive duplications or repeats of codons. Selective immunoreactivity of different parts of the dystrophin molecule in sections of muscle biopsy material distinguishes the Duchenne and Becker forms (Fig. 601-3). The demonstration of deletions and duplications also can be made from blood samples by the more rapid PCR, which identifies as many as 98% of deletions by amplifying 18 exons but cannot detect duplications. The diagnosis can thus be confirmed at the molecular genetic level from either the muscle biopsy material or from peripheral blood, although as many as 30% of boys with DMD or BMD have a false-normal blood PCR; all cases of dystrophinopathy are detected by muscle biopsy.
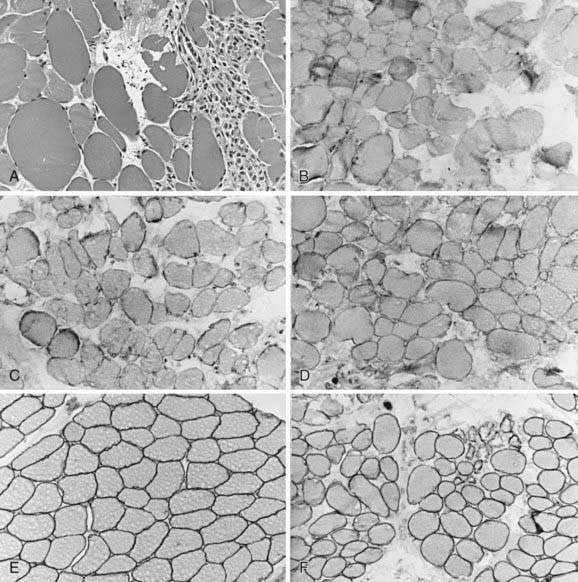
Figure 601-3 Quadriceps femoris muscle biopsy specimens from a 4 yr old boy with Becker muscular dystrophy. A, Myofibers vary greatly in size, with both atrophic and hypertrophic forms; at the right is a zone of degeneration and necrosis infiltrated by macrophages, similar to Duchenne muscular dystrophy (H&E, ×250). Immunoreactivity using antibodies against the dystrophin molecule in the rod domain (B), carboxyl-terminus (C), and amino-terminus (D) all show deficient but not totally absent dystrophin expression; most fibers of all sizes retain some dystrophin in parts of the sarcolemma but not around the entire circumference in cross section. Alternatively, the prominence of dystrophin is less, appearing weak, when compared with the simultaneously incubated normal control from another child of similar age (E). F, Merosin expression is normal in this patient with Becker dystrophy, in both large and small myofibers, and is lacking only in frankly necrotic fibers. Compare with classic Duchenne muscular dystrophy illustrated in Figure 601-2C and with Figure 601-5.



