Molecular Pathogenesis of Gynecologic Cancers
ANDREW BERCHUCK  DOUGLAS A. LEVINE
DOUGLAS A. LEVINE  JOHN H. FARLEY
JOHN H. FARLEY  MICHAEL J. BIRRER
MICHAEL J. BIRRER
INTRODUCTION
An overview of the molecular pathogenesis of cancer is presented in the initial sections of this chapter. A more comprehensive discussion of this topic can be found in Part I of DeVita Hellman and Rosenberg’s Cancer: Principles and Practice of Oncology (1). Genetic alterations involved in the development of gynecologic cancers are covered in more detail in the latter sections of this chapter.
The causes of human cancers are diverse, but malignant transformation is invariably caused by the development of genetic alterations that disrupt cell growth (Table 2.1). Several classes of genetic alterations are involved in carcinogenesis, including changes in the sequence of genes (mutations), gains (amplifications) or losses (deletions) in the number of copies of genes and rearrangement and translocation of genes from their normal chromosomal locations (2). These alterations lead to activation of genes that stimulate proliferation (oncogenes) and inactivation of genes that inhibit proliferation (tumor-suppressor genes). Disruption of DNA-repair systems also often occurs in the process of malignant transformation and may lead to accelerated accumulation of genetic alterations. Because repair of genetic damage inhibits carcinogenesis, DNA-repair genes are also considered tumor suppressors.
Most transforming mutations in oncogenes alter a single amino acid at specific codons that produce overactive protein products (e.g., KRAS, BRAF), and only one allele need be mutated. In contrast, inactivation of tumor suppressors (e.g., RB1, BRCA1) requires loss of both copies of the gene (two hits). Mutations in tumor suppressors occur throughout the gene, and are usually small insertions, deletions or base substitutions that result in truncated protein products. Loss of the second nonmutated allele generally occurs due to chromosomal deletion, leading to abrogation of tumor-suppressor activity.
Cancers may have many genetic alterations, but only a small fraction of these are “driver” mutations responsible for malignant transformation. The majority are “passenger” mutations that arise secondarily due to genetic instability. Although not involved in the initial monoclonal development of a cancer, passenger mutations may play a role in clonal evolution and progression of the malignant phenotype. This may alter proteases involved in invasion and metastasis and caspases that regulate cell death in response to therapy.
Changes in gene expression due to epigenetic alterations (DNA methylation and acetylation), aberrant gene splicing, noncoding microRNAs, and other mechanisms also contribute to the malignant phenotype, but sequential genetic alterations leading to outgrowth of a clonal population of cells is the major determinant of whether a cancer develops as well as the time required for its development and progression. The histopathologic progression through hyperplasia, dysplasia, carcinoma in situ, and invasive carcinoma, reflects the multistep, multigenic nature of malignant transformation. Cancers may arise through the accumulation of acquired (somatic) alterations or through the inheritance of an alteration in the germline followed by the acquisition of additional somatic alterations (Fig. 2.1). Hereditary cancers are discussed in detail in Chapter 3.
Molecular Alterations in Cancer |
Genetic changes
Mutations
Mutations in oncogenes change a single amino acid and lead to gain of activity that stimulates proliferation (e.g., KRAS). These mutations are dominant and not dependent on changes in the other copy of the gene.
Mutations in tumor-suppressor genes (e.g., PTEN) and DNA repair genes (e.g., BRCA1) generally are small base insertions/deletions or single base changes that cause stop codons. These lead to truncated protein products that are nonfunctional. Loss of tumor-suppressor activity is usually dependent on deletion of the other copy of the gene.
Driver mutations play a critical role in the process of malignant transformation
Passenger mutations occur due to generalized genomic instability. Although not involved in malignant transformation, they may contribute to cancer phenotypes (e.g., metastatic potential, response to therapy)
DNA copy number changes
Deletion of one or both copies of a tumor-suppressor gene due to genomic instability leads to loss of a gene product (e.g., RB1)
Gain of additional copies of an oncogene leads to increased activity (e.g., HER-2/neu)
Gene rearrangements and translocations
When a gene is moved from its normal location to another chromosomal location, its expression may be increased due to proximity to a gene promoter (e.g., ALK in lung cancer)
Aneuploidy
Aneuploidy with gain or loss of complete chromosomes is common in many cancers (e.g., high-grade serous ovarian cancer) and likely plays a role in their development and progression.
Other biological processes
Epigenetic alterations
Hypermethylation of CpG islands in the promoter regions of tumor-suppressor genes may lead to gene inactivation (e.g., MLH1).
Loss of promoter methylation in genes that stimulate proliferation may provide an oncogenic stimulus (e.g., IGF2).
Changes in acetylation of histone proteins that coat DNA may also play a role in carcinogenesis.
Aberrant gene splicing
Genes may have alternative splice forms that produce messenger RNAs and proteins that have altered activity.
MicroRNAs
These small noncoding RNAs can affect gene expression by binding to messenger RNAs and blocking their translation. Altered miRNA expression plays a role in the development and behavior of some cancers.
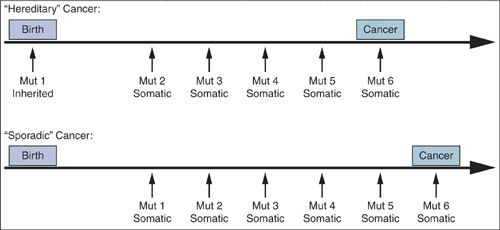
FIGURE 2.1. All cancers are genetic. “Hereditary” cancers differ from “sporadic” cancers by virtue of association with a predisposing mutation inherited through the germ line. In contrast, all of the mutations in sporadic cancers are acquired somatically.
The techniques used to study genetic alterations have evolved dramatically. Advances in the 1980s and 1990s enabled the discovery of several genes that frequently play a role in the development of cancers, including the KRAS and HER-2/neu oncogenes and the TP53 and RB1 tumor-suppressor genes. It became apparent that the spectrum of driver alterations varies considerably between cancer types, and among cancers of the same type. More recently, powerful new technologies have enabled comprehensive genomic studies. The Human Genome Project was completed in 2004 and provided a road map of the entire genome, including the approximately 18,000 protein-encoding genes. The HapMap project then catalogued the millions of common single nucleotide polymorphisms (SNPs) in the genome that change a single base in the DNA sequence. More recently the 1000 Genomes Project has characterized rarer germline genetic variants. A road map of normal human genetic variation and the development of faster and cheaper “next generation” DNA sequencing has allowed mutational analysis of complete cancer genomes. It has been shown that cancers typically have about 30 to 100 acquired mutations. However, cancers are continually evolving due to genetic instability and contain subclones that differ with respect to their mutational profile. A small number of genes are mutated in a high fraction of cancers (e.g., KRAS, TP53) likely reflecting the strong selective advantage these alterations confer. A much larger number of genes are mutated at considerably lower frequencies. The terms “mountains” and “hills” have been used to describe genes that are commonly versus rarely mutated. Mutations in cancers typically target multiple biological pathways (3). Several genes in a pathway may be mutated, but driver mutations in a pathway often are mutually exclusive when they involve the same pathway (e.g., KRAS and BRAF).
The Cancer Genome Atlas (TCGA) Project sponsored by the National Cancer Institute is performing a comprehensive genomic characterization of all common forms of human cancer. Ovarian and endometrial cancers were among the first to be studied by TCGA and these findings have been incorporated into this chapter. The International Cancer Genome Consortium (ICGC) is performing similar genomic analyses of human cancers. Both TCGA and ICGC are depositing their data online in the public domain to stimulate further research. The full spectrum and complexity of the genomic alterations in a single cancer can be best captured in a Circos plot (Fig. 2.2), which uses a circular ideogram layout to facilitate the display of changes across the genome.
An understanding of the molecular pathogenesis of cancer provides the opportunity to better define subgroups within a given type of cancer that may differ with respect to clinical behavior or outcome. In addition, the identification of specific mutations has facilitated the development of therapies that target these alterations (4,5). In this regard, monoclonal antibodies and small molecules targeting aberrantly expressed cellular proteins that drive malignant growth have been successful in treating some cancers (e.g., trastuzumab targets HER-2/neu in breast cancer, gefitinib, and erlotinib target EGFR in lung and other cancers). However, resistance to therapy often occurs due to mutational heterogeneity both within the primary tumor and in metastases as clonal evolution develops over time. Unfortunately, many cancers, including most gynecologic cancers, have not proven easily amenable to targeted therapy, perhaps because their growth is not driven by a single altered gene or pathway. Successful therapy may require the use of multiple agents that target more than one gene or pathway.
In several types of common cancers, analysis of known “druggable” molecular targets has become part of the standard of care. Companion molecular diagnostic tests are often requisite to guide selective use of expensive targeted agents in patients most likely to benefit. As the cost of genomic analyses of cancers continues to fall, it will be increasingly feasible to perform full sequencing of cancers to look for less commonly mutated targets. This has the potential to facilitate even greater individualization of therapy based on the unique profile of alterations in a cancer, but the utility of this approach remains unproven.
Finally, an increased understanding of the molecular pathogenesis provides the opportunity to explore new approaches to early detection and prevention. This has been realized for cervical cancer with the identification of the human papilloma virus leading to creation of a vaccine. In ovarian and endometrial cancers, genetic changes that occur early in the development of cancers have the potential to serve as biomarkers for early detection and as surrogate endpoints in prevention studies.
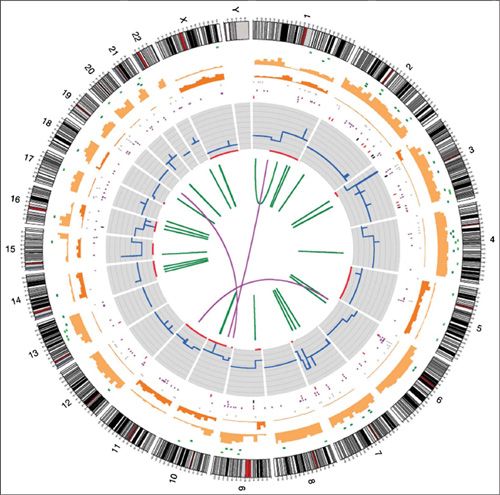
FIGURE 2.2. Circos plot of somatic mutations in COLO-829. Chromosome ideograms are shown around the outer ring and are oriented pter–qter in a clockwise direction with centromeres indicated in red. Other tracks contain somatic alterations (from outside to inside): validated insertions (light green rectangles); validated deletions (dark green rectangles); heterozygous (light orange bars), and homozygous (dark orange bars) substitutions shown by density per 10 megabases; coding substitutions (colored squares: silent in gray, missense in purple, nonsense in red, and splice site in black); copy number (blue lines); regions of loss of heterozygosity (LOH) (red lines); validated intrachromosomal rearrangements (green lines); validated interchromosomal rearrangements (purple lines).
Source: Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 2010;463(7278):191. [PMID: 20016485]
CELLULAR GROWTH AND DEATH
Proliferation
The number of cells in normal tissues is tightly regulated by a balance between cellular proliferation and death. The final common pathway for cell division involves distinct molecular switches that control cell cycle progression from G1 to the S phase of DNA synthesis (6). These include the Rb and E2F proteins and their various regulatory cyclins, cyclin dependent kinases (cdks) and cdk inhibitors. The events that facilitate progression from G2 to mitosis and cell division are regulated by other cyclins and cdks (Fig. 2.3), as well as by molecules involved in chromosomal segregation such as microtubules. When the chromosomes do not separate properly, aneuploidy may occur with gain or loss of complete chromosomes (7). Aneuploidy is a hallmark of many cancers and likely plays a role in their development and progression.
Cell cycle progression is also dependent on ubiquitin-mediated proteolysis of molecules involved in cell cycle arrest (6). Ubiquitin is a 76 amino acid polypeptide that can be linked to lysine residues of proteins, and ubiquitination marks proteins for proteolysis and destruction by the proteasome complex. Proteosome inhibitors have been successfully employed to treat some hematologic malignancies, and the FBXW7 (CDC4) ubiquitin ligase has been shown to be mutated in some serous uterine cancers.
In some tissues—such as the bone marrow, epidermis, and gastrointestinal tract—the life span of mature cells is relatively short and high rates of proliferation are required to maintain the population; while in other tissues—such as liver, muscle and brain—cells are long lived and proliferation rarely occurs. Complex molecular mechanisms have evolved to closely regulate proliferation. These involve a finely tuned balance between growth stimulatory and inhibitory signals.
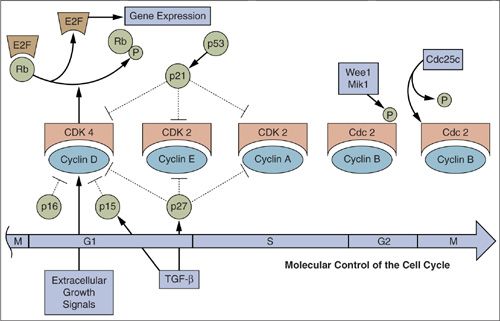
FIGURE 2.3. Molecular control of cell-cycle progression. A linear version of the various stages of the cell cycle is shown with the various cyclin/cyclin-dependent kinase complexes corresponding to the stages that they control.
Increased proliferation is one of the hallmarks of cancer. There may be increased activity of genes involved in stimulating proliferation (oncogenes) and/or loss of growth-inhibitory (tumor-suppressor) genes. In the past, it was thought that cancer might arise solely because of more rapid proliferation or a higher fraction of cells proliferating. It is now clear that this was an overly simplistic view. Although increased proliferation is a characteristic of many cancers, the fraction of cancer cells actively dividing and the time required to transit the cell cycle is not always strikingly increased. Altered regulation of proliferation is only one of several factors that contribute to malignant transformation.
Cell death
In addition to being driven by increased proliferation, growth of a cancer may be attributable to cellular resistance to death. At least three distinct types of cell death pathways have been characterized, including apoptosis, autophagy, and necrosis (8).
Apoptosis
The term “apoptosis” derives from Greek and alludes to a process akin to leaves dying and falling off a tree. Apoptosis is an active energy-dependent process that involves cleavage of the DNA by endonucleases and proteins by proteases called caspases. Morphologically, apoptosis is characterized by condensation of chromatin, nuclear and cytoplasmic blebbing, and cellular shrinkage. The molecular events that effect apoptosis in response to various stimuli are complex and have only been partially elucidated, but several reliable markers of apoptosis have been discovered including annexin V, caspase-3 activation, and DNA fragmentation.
External stimuli such as TNF, TRAIL, Fas and other death ligands that interact with cell surface receptors can induce activation of caspases and lead to apoptosis via the extrinsic pathway. The intrinsic apoptosis pathway is activated in response to a wide range of stresses including DNA damage and deprivation of growth factors. The intrinsic apoptosis pathway is regulated by a complex interaction of pro- and antiapoptotic proteins in the mitochondrial membrane that affect its permeability. Proteins that increase permeability allow release of cytochrome c, which activates the apoptosome complex leading to activation of caspases that effect apoptosis. Conversely, proteins that stabilize mitochondrial membranes inhibit apoptosis. The first major insight that led to the understanding of the intrinsic apoptosis pathway was the finding that an activating translocation of the BCL-2 gene in B-cell lymphomas results in essentially complete inhibition of apoptosis (9). Subsequent studies demonstrated that the antiapoptotic effect of BCL-2 is attributable to stabilization of the mitochondrial membrane. Additional genes related to BCL-2, such as BAD, BCL-XL and others, also block apoptosis by inhibiting membrane permeability. Other genes in the BCL family, such as BAX and BAK, increase membrane permeability and are pro-apoptotic. An understanding of the complex system of checks and balances involved in regulation of apoptosis provides opportunities for the development of targeted therapies (10).
In addition to restraining the number of cells in a population, apoptosis serves an important role in preventing malignant transformation by allowing for elimination of cells that have undergone genetic damage. Following exposure of cells to mutagenic stimuli, including radiation and carcinogenic drugs, the cell cycle is arrested so that DNA damage may be repaired. If DNA repair is not sufficient, apoptosis occurs so that damaged cells do not survive. This serves as an anticancer surveillance mechanism by which mutated cells are eliminated before they become fully transformed. The TP53 tumor-suppressor gene is a critical regulator of cell cycle arrest and apoptosis in response to DNA damage. Many of the molecules involved in apoptosis reside in the mitochondria and are encoded by mitochondrial DNA. Mutations in mitochondrial DNA have been shown to be frequent in cancer and may play a role in evasion of apoptosis (11).
Necrosis
Necrosis is a type of cell death that is distinct from apoptosis and is the result of bioenergetic compromise (12). Morphologic changes include swollen organelles and rupture of the cell membrane leading to loss of osmoregulation and cellular fragmentation. Necrosis is a less well-regulated process that leads to spillage of protein contents and this may incite a brisk immune response. This is in contrast to the silent elimination of cells by apoptosis, which typically elicits a minimal immune response. There is evidence that some drugs may enhance necrotic death in tumors and this may stimulate a beneficial antitumor immune response.
Autophagy
Autophagy is a potentially reversible process in which a cell that is stressed “eats” itself (13). A wide range of stresses have been identified that may elicit autophagy (some of which may also elicit apoptosis), including growth factor deprivation and accumulation of reactive oxygen species. Unlike necrosis and apoptosis, in which loss of integrity of the cytoplasmic and nuclear membranes respectively are defining events, autophagy is characterized by the formation of cytoplasmic autophagic vesicles into which cellular proteins and organelles are sequestered. This may allow for cell survival if damaged organelles can be repaired. Conversely, the process may lead to cell death if these vesicles fuse with lysosomes with resultant degradation of their contents. Several cancer therapeutic agents have been shown to induce autophagy, while targeted disruption of genes such as ATG5 that are involved in autophagy can inhibit cell death.
Cellular Senescence
Normal cells are only capable of undergoing division a finite number of times before becoming senescent and this represents a barrier to immortality (14). Senescence can also be triggered by stresses such as oncogene activation. Induction of senescence is mediated by persistent p16 and p53 activation leading to permanent cell cycle arrest. Cellular senescence is regulated by a biological clock related to progressive shortening of repetitive DNA sequences (TTAGGG) called telomeres that cap the ends of chromosomes. Telomeres are involved in chromosome stabilization and in preventing recombination and chromosomal translocations and aneuploidy during mitosis (15). At birth, chromosomes have long telomeric sequences (150,000 bases) that become progressively shorter by 50 to 200 bases each time a cell divides. Telomerase is a ribonucleoprotein complex. The RNA component serves as a template for telomere extension and the protein subunit acts to catalyze the synthesis of new telomeric repeats.
Replicative senescence serves as a defense against immortalization and malignant transformation by placing limits on proliferation of cells that have accumulated genetic damage. Reactivation of telomerase is critical to the emergence of cancers (16), but early in the process of malignant transformation telomere shortening and dysfunction may promote the carcinogenic process through the generation of chromosomal rearrangements and aneuploidy (17). Progressive telomere shortening during adulthood may explain the association between advancing age and increased cancer risk, particularly for epithelial malignancies. This risk has been mainly attributed to progressive accumulation of mutations with aging, but this theory does not explain the marked aneuploidy of most epithelial cancers.
Telomerase activity is present in a high fraction of many cancers, including ovarian (18), cervical (19) and endometrial cancers (20). It has been suggested that detection of telomerase might be useful for early diagnosis of cancer, but lack of specificity is a significant issue. In this regard, endometrium is one of the normal adult tissues in which telomerase expression is most common (21). Perhaps this relates to the need for a large number of lifetime cell divisions because of rapid growth and shedding of this tissue each month during the reproductive years. Therapeutic approaches to inhibiting telomerase are under development that would reverse their immortalized state and render them susceptible again to normal replicative senescence (22).
Stem Cells
Stem cells in normal tissues have the capacity to undergo asymmetric division to produce daughter cells that undergo differentiation and eventually senescence, and can self-renew to produce more stem cells (23). Stem cells have been most easily identifiable in tissues that normally are highly proliferative, such as the bone marrow, skin and intestine, in which a constant stream of cells are being born to replace those that are differentiating and dying. It has been theorized that stem cells may exist in cancers and that these have the capacity for self-renewal and differentiation into populations of cells that recapitulate tumor heterogeneity (24). Stem cells are also referred to as tumorigenic cells because of their capacity to regenerate tumors. The stem cell theory suggests that less than 1% of cells in a cancer are stem cells and that by virtue of being dormant or quiescent, they may be relatively resistant to chemotherapy and radiation. It is postulated that cancer stem cells may persist as microscopic disease after eradication of most cancer cells and give rise to recurrent disease. There has been a great deal of interest in identifying and characterizing cancer stem cells, as this could facilitate development of therapies that specifically target these resistant cells.
Several molecular markers have been identified that define cancer stem cells (25), including the cell membrane glycoprotein CD133 (Prominin). Expression of CD133 has been associated with greater proliferative potential and ability to form tumors in animal models. CD44 is the receptor for hyaluronic acid and has been identified as a stem cell marker in several cancer types. CD117, also known as c-kit, is another well-characterized stem cell marker that has been implicated in several solid tumors (25). CD24 is a mucin-like cell surface glycoprotein marker that has been identified as a stem cell marker. The Sonic Hedgehog and WNT pathways also have been implicated in stem cell behavior.
Although expression of the markers and pathways described above in cancers have been associated with stem cell characteristics, the existence of discrete cancer stem cell populations has not been definitively proven. It is possible that the genes and pathways involved in cellular renewal are simply more highly expressed in a fraction of cells within heterogeneous tumors (26). Cancer stem cells may exist in some malignancies but not others. In this regard, demonstration of progenitor stem cells has been more robust in leukemias than in solid tumors.
ORIGINS OF GENETIC ALTERATIONS
Human cancers arise due to a series of genetic alterations that lead to disruption of normal mechanisms that govern cell growth, death, and senescence (2). Genetic damage may be inherited or arise after birth due to various exposures including those related to carcinogenic substances such as tobacco and asbestos, physical factors such as ultraviolet radiation, infections such as HPV, dietary factors, obesity, and inflammation. However much of the acquired genetic damage that causes cancer is due to endogenous mutagenic processes within the cell (Table 2.2). The incidence of most cancers increases with aging because the longer a person is alive the higher the likelihood of a cell acquiring sufficient damage to become fully transformed. It is thought that at least three to six alterations are required to fully transform a cell. Many cancers are genetically unstable and this leads to accumulation of a substantial number of secondary changes and the development of subclones that play a role in evolution of the malignant phenotype.
Origins of Genetic Damage in Human Cancers |
Type of genetic damage | Examples |
Germline alterations |
|
High-penetrance genes | BRCA1, BRCA2 (breast, ovarian cancers) MLH1, MSH2 (Lynch syndrome) |
Low-penetrance genes | SNPs associated with various cancers |
Exogenous carcinogens |
|
Ultraviolet radiation | TP53 mutations in skin cancers |
Tobacco | TP53 mutations in lung cancers |
Viruses | HPV inactivation of RB and TP53 in cervical cancers |
Endogenous DNA damage |
|
Cytosine methylation and deamination | TP53 mutations in many cancer types |
DNA Hydrolysis | various genes |
Spontaneous errors in DNA synthesis | various genes |
Free radical production due to oxidative stress | various genes |
Inherited Cancer Susceptibility
Although most cancers arise sporadically due to acquired genetic damage, inherited mutations in cancer susceptibility genes are responsible for some familial cases. The age of cancer onset is younger in these families and it is not unusual for some individuals to be affected with multiple primary cancers. Tumor-suppressor genes and DNA repair genes have been implicated most frequently in hereditary cancer syndromes. The most common forms of hereditary cancer syndromes predispose to breast/ovarian (BRCA1/2 genes) and colon/endometrial (DNA mismatch repair genes) cancers. Although affected individuals carry the germline alteration in every cell of their bodies, paradoxically, cancer susceptibility genes are characterized by a limited repertoire of cancers. The penetrance of cancer susceptibility genes is incomplete, as all individuals who inherit a mutation do not develop cancer. The emergence of cancers is dependent on the occurrence of additional genetic alterations. Hereditary cancer syndromes are covered in detail in Chapter 3.
In addition to the high-penetrance mutations noted above that cause familial cancer syndromes, low-penetrance genetic variants may also affect cancer susceptibility, albeit less dramatically (27). There are over 10 million polymorphic genetic loci in the human genome, most of which are SNPs (e.g., CCA to CAA). Many of these SNPs are common, with the rarer of the two alleles occurring in more than 5% of individuals. Genome wide association studies have discovered a number of SNPs that affect the risk of ovarian cancer by 10% to 20% per rare allele copy (27). A more complete understanding of the genetic factors that affect cancer susceptibility could facilitate implementation of screening and prevention approaches in subsets of the population at increased risk.
Acquired Genetic Damage
The etiology of acquired genetic damage in cancers also has been elucidated to some extent. For example, a strong causal link exists between cigarette smoke and cancers of the aerodigestive tract, between ultraviolet radiation and skin cancer, and between HPV and lower genital tract cancers (Table 2.2). For many common forms of cancer (e.g., colon, breast, prostate, endometrium, ovary), there is not a strong association with specific carcinogens. It is thought that the genetic alterations responsible for these cancers may arise mainly due to endogenous mutagenic processes. This includes methylation and deamination of cytosine residues leading to transition mutations to thymidine. In this regard, in most types of cancers that are not associated with a specific carcinogen, transition mutations from purine to purine or pyrimidine to pyrimidine are much more common than transversion mutations that change purine to pyrimidine or pyrimidine to purine. An additional mechanism of endogenous damage includes spontaneous errors in DNA synthesis that frequently occur during the process of DNA replication associated with normal proliferation. Free radicals generated in response to inflammation and other cellular damage may also cause DNA damage. These endogenous processes are thought to produce mutations on an ongoing basis. Several networks of highly effective DNA damage surveillance and repair genes exist, but some mutations may elude them. The efficiency of these DNA damage response systems varies between individuals due to genetic and other factors and may affect susceptibility to cancer.
Epigenetic Changes
Epigentics comprises heritable changes that are not due to alterations in DNA sequence (28,29). Methylation of cytosine residues that reside next to guanine residues (CpG dinucleotides) is the primary mechanism of epigenetic regulation, and this process is regulated by a family of DNA methyltransfereases. CpG dinucleotides are asymmetrically distributed with about half of human genes containing CpG-rich regions termed “CpG islands” at their transcriptional start sites. Most genes are regulated without changing the methylation status of the CpG sites, but permanent silencing of genes associated with X-chromosome inactivation and genomic imprinting is due to heritable methylation of CpG islands.
Most cancers have globally reduced DNA methylation, which may lead to activation of some genes. Conversely, selective hypermethylation of CpG islands in the promoter regions of tumor-suppressor genes may lead to gene inactivation (e.g., BRCA1, MLH1). In addition, loss of silencing of imprinted genes that stimulate proliferation, such as IGF2, may provide an oncogenic stimulus. Acetylation and methylation of the histone proteins that coat DNA represent another level of epigenetic regulation that is altered in cancer. The underlying cause of epigenetic alterations in cancer remains poorly understood.
Alterations in Signal Transduction Pathways in Malignant Transformation
Alterations in genes that stimulate cellular growth (oncogenes) can cause malignant transformation when they become overactive (2). In some cancers, amplification of oncogenes occurs with resultant overexpression of the corresponding protein. Instead of two copies of one of these genes, there may be many additional copies. Some oncogenes may become overactive when affected by gain of function point mutations. Finally, oncogenes may be translocated from one chromosomal location to another and come under the influence of gene promoters that cause overexpression. This latter mechanism frequently occurs as a driving event in the development of leukemias and lymphomas, but is infrequent in gynecologic cancers. However, some other types of solid tumors may have translocations that represent significant driver events, such as those involving the ALK receptor gene in lung cancer. These have been targeted successfully with the monoclonal antibody crizotinib (30).
Loss of tumor-suppressor gene function also plays a role in the development of most cancers. This usually involves a two-step process in which both copies of a tumor-suppressor gene are inactivated. This often involves mutation of one copy of a tumor-suppressor gene and loss of the other copy due to deletion of a segment of the chromosome where the gene resides (e.g., RB1). There is also evidence that some tumor-suppressor genes may be inactivated due to methylation of the promoter region of the gene (e.g., MLH1, BRCA1). The promoter is an area proximal to the coding sequence that binds transcription factors that regulate whether or not the gene is transcribed from DNA into RNA. When the promoter is methylated, it is resistant to activation and the gene is essentially silenced despite remaining structurally intact. This two-hit paradigm of tumor-suppressor gene inactivation is relevant to both hereditary cancer syndromes, in which one mutation is inherited and the second acquired, and to sporadic cancers, in which both hits are acquired. Tumor-suppressor gene products are found throughout the cell, reflecting their diverse functions.
The sections below will review the role of oncogenes and tumor-suppressor genes in signal transduction pathways that regulate cellular growth and metabolism (3). These pathways are complex and have considerable overlap and redundancy (31), and an exhaustive discussion is beyond the scope of this book. Pathways for which there is less evidence of a role in driving the development of gynecologic cancers, such as the Hedgehog, Notch, and cytokine pathways, are not covered.
Peptide Growth Factors and Receptor Tyrosine Kinases
Peptide growth factors in the extracellular space, such as those of the EGF, PDGF, and FGF families, stimulate a cascade of molecular events that leads to proliferation by binding to high affinity cell membrane receptors (Fig. 2.4). Growth factors are involved in normal cellular processes such as stromal-epithelial communication, tissue regeneration, and wound healing. The concept that autocrine growth stimulation might be a key strategy by which cancer cell proliferation becomes autonomous has received considerable attention. In this model, it is postulated that cancers secrete stimulatory growth factors that then interact with receptors on the same cell. Although peptide growth factors provide a growth stimulatory signal, there is little evidence to suggest that overproduction of growth factors is a precipitating event in the development of most cancers. Increased expression of peptide growth factors likely facilitates, rather than drives, malignant transformation.
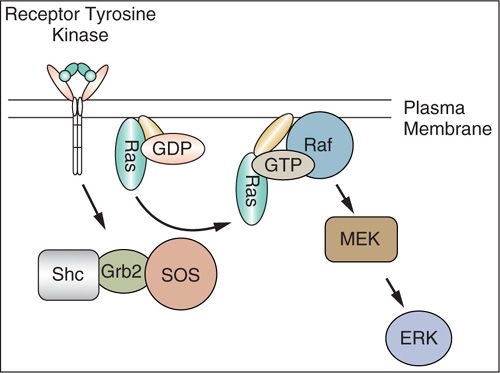
FIGURE 2.4. Receptor tyrosine kinase pathway. Most receptor tyrosine kinases stimulate the activity of the Ras guanine nucleotide exchange factor son of sevenless (SOS), which associates with the linker proteins Shc and Grb2. The activation of Ras by SOS stimulates a protein serine kinase cascade initiated by Raf, which stimulates MEK. MEK then activates the extracellular signaling-regulated kinases (ERKs). ERKs phosphorylate transcription factors to regulate gene expression.
Cell membrane receptors that bind peptide growth factors are composed of an extracellular ligand binding domain, a membrane-spanning region, and a cytoplasmic tyrosine kinase domain (31,32). Binding of a growth factor to the extracellular domain results in dimerization and conformational shifts in the receptors and activation of the inner tyrosine kinase. The kinase transfers a phosphate group from ATP to specific tyrosine residues both on the growth factor receptor itself (autophosphorylation) and on molecular targets in the cell interior leading to activation of secondary signals that stimulate proliferation. Growth of some cancers is driven by overexpression of receptor tyrosine kinases. The epidermal growth factor receptor (EGFR) family of receptor tyrosine kinases plays a significant role in the development of several types of cancers, and includes ErbB-1 (EGFR), ErbB-2 (HER-2/neu), ErbB-3, and ErbB-4. These receptors are activated by binding of ligands, including EGF, transforming growth factor-α, amphiregulin, and the neuregulins.
Because receptor tyrosine kinases are located on the cell surface, they are appealing therapeutic targets. A number of agents that target the EGFR family have been developed and translated into clinical practice. Trastuzumab is a monoclonal antibody that binds to HER-2/neu, and it is widely used in the treatment of breast cancers that overexpress this receptor (33). Cetuximab is a monoclonal antibody that targets the extracellular domain of EGFR, whereas gefitinib is a direct inhibitor of the EGFR tyrosine kinase (34). Lapatinib is a dual EGFR/HER-2 kinase inhibitor. Likewise, therapeutic approaches have been developed that target other receptor tyrosine kinases. Imatinib antagonizes the activity of the BCR-ABL, c-kit, and platelet-derived growth factor (PDGF) receptor tyrosine kinases and has proven highly effective in treatment of chronic myelogenous leukemias and gastrointestinal stromal tumors.
Nonreceptor Kinases and Phosphatases
Following interaction of growth factors with cell membrane receptors, secondary molecular signals are generated to transmit the growth stimulus to the nucleus. This function is served by a multitude of complex and overlapping signal transduction pathways that occur in the inner cell membrane and cytoplasm. Many of these signals involve phosphorylation of proteins by enzymes known as nonreceptor kinases. The kinases that are involved in growth regulation are of two types, those that phosphorylate tyrosine residues of target proteins, including those of the SRC family (35), and others that are specific for serine and/or threonine residues such as AKT (36). The activity of kinases is opposed by phosphatases such as PTEN, which act in opposition to the kinases by removing phosphates from the target proteins. The PTEN gene is among the most frequently mutated tumor-suppressor genes in human cancers.
RAS/RAF, Mitogen-Activated Protein Kinase (MAPK) Pathway
Guanosine-triphosphate–binding proteins (G proteins) such as RAS represent another class of molecules involved in transmission of growth signals (Figs. 2.4 and 2.5A). They are located on the inner aspect of the cell membrane and are positively regulated by Grb and SOS in response to receptor tyrosine kinases and other signals. They have intrinsic GTPase activity that catalyzes the exchange of GTP (guanine-tri-phosphate) for GDP (guanine-di-phosphate). In their active GTP bound form, G proteins interact with kinases that are involved in relaying the mitogenic signal, such as those of the MAP kinase family. Conversely, hydrolysis of GTP to GDP, which is stimulated by GTPase activating proteins (GAPs), leads to inactivation of G proteins. The RAS family of G proteins is among the most frequently mutated oncogenes in human cancers. Activation of RAS genes usually involves point mutations in codons 12, 13, or 61 that result in constitutively activated molecules (37).
The RAF family of genes encode serine-threonine kinases that interact with RAS proteins and propagate signaling by activating MAP kinases (MEK) which translocate to the nucleus. Mutations in the BRAF gene occur in many cancers that lack RAS mutations and most of these mutations involve codon 599 in the kinase domain (38). The mutual exclusivity of KRAS and BRAF mutations is consistent with the need to only activate one gene in a pathway. Melanomas often have mutations in codon 600 of BRAF. The BRAF kinase inhibitor vemurafenib has been shown to be highly active in melanomas with the BRAF V600E mutation (39).
The RAF serine/threonine kinase is downstream of RAS, and its activation leads to signaling via mitogen-activated protein kinase kinase (MAPKK), also called MEK, which activates mitogen-activated protein (MAP) kinase or extracellular regulated kinase (ERK). MAP kinase activation results in phosphorylation and activation of nuclear factors such as ribosomal S6 kinase and transcription factors JUN, MYC and FOS, resulting in the switching on of a number of genes associated with proliferation.
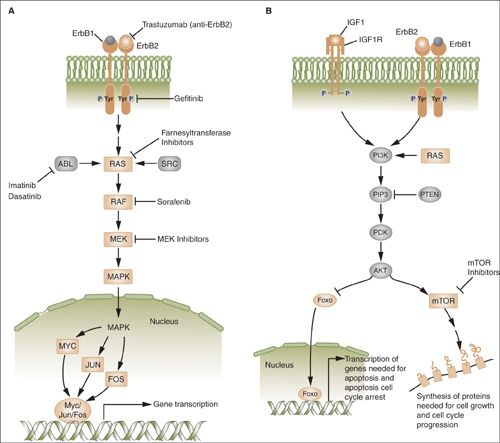
FIGURE 2.5. A: The ras/raf/MEK/MAPK pathway is activated by multiple growth factor receptors (here exemplified by ErbB1 and ErbB2) as well as several intracellular tyrosine kinases such as SRC and ABL. Activated RAS stimulates a sequence of phosphorylation events mediated by RAF, MEK, and ERK (MAP) kinases. Activated MAP kinase (MAPK) translocates to the nucleus and activates proteins such as MYC, JUN, and FOS that promote the transcription of numerous genes involved in tumor growth. B: The phosphatidylinositol 3-kinase (PI3K) pathway is activated by RAS and by a number of growth factor receptors (here exemplified IGF1R and the ErbB1/ErbB2 heterodimer). Activated PI3K generates phosphatidylinositol-3,4,5-triphosphate (PIP3), which activates phosphoinositide-dependent kinase-1 (PDK). In turn, PDK phosphorylates AKT. PTEN is an endogenous inhibitor of AKT activation. Phosphorylated AKT transduces multiple downstream signals, including activation of the mTOR and inhibition of the FOXO family of transcription factors. mTOR activation promotes the synthesis of proteins required for cell growth and cell cycle progression.
PI3K/AKT/mTOR Pathway
Phosphatidylinositol 3-kinases (PI3Ks) are a class of lipid kinases that phosphorylate phosphoinositides (PIs) at the position D3 of the inositol ring (40). Extracellular molecules such as growth factors are the main effectors for PI3K pathway activation through the interaction with receptor tyrosine kinases and G protein-coupled receptors (Fig. 2.5B). Phosphorylated PIs serve as plasma docking sites for the recruitment of pleckstrin-homology domain containing proteins such as AKT and its upstream activator PDK1. PI3K activation is one of the main causes of increased proliferation and resistance to apoptosis. The PIK3CA gene is one of the most frequently mutated oncogenes in human cancers.
Alterations in PI3Ks and downstream effectors, including AKT and mTOR, frequently are involved in initiation and maintenance of the tumorigenic phenotype. The PI3K-AKT pathway promotes cell growth and survival and inhibits apoptosis and autophagy through several mechanisms. AKT activates the mammalian target of rapamycin (mTOR) pathway (Fig. 2.5B) and modulates genes that inhibit cell cycle progression (e.g., cdks, CHK1, and MDM2). AKT targets include the pro-apoptotic effectors BAD and BAX, which are inhibited by phosphorylation respectively at Ser136 and Ser184, resulting in inhibition of the apoptotic response. AKT also inhibits the expression of BH3-only proteins such as BAD and BIM through effects on transcription factors, such as Forkhead family proteins (e.g., FOXO3a) and p53. AKT also influences p53 activity through MDM2 phosphorylation at Ser166 and Ser186, which promotes its translocation to the nucleus, with subsequent destabilization. AKT can phosphorylate and activate IκB kinase-α, which in turn phosphorylates IκB, targeting it for degradation. This leads to nuclear translocation and activation of the transcription factor NF-κB and transcription of NF-κB–dependent pro-survival genes.
The PTEN tumor suppressor is the most important negative regulator of the PI3K signaling pathway (41). It is a lipid and protein phosphatase that removes phosphates in opposition to PI3K tyrosine kinases. Its ability to dephosphorylate phosphatidylinositol 3,4,5-trisphosphate (PIP3) resulting in phosphatidylinositol 4,5-bisphosphate (PIP2) inhibits oncogenic PI3K-dependent signaling. Although PTEN is a tumor suppressor and may be completely lost due to mutation and deletion of the two copies of the gene, mutational inactivation of one copy of the gene (haploinsufficiency) also appears to facilitate malignant transformation.
Both genetic and epigenetic alterations affect the activity of this pathway (40). Mutations in the PIK3CA gene that lead to increased activity frequently occur in human cancers, including ovarian and endometrial cancers. PTEN is frequently targeted by either germline or somatic mutation, and is the second most frequently mutated gene in human cancers. In addition, loss of heterozygosity or promoter hypermethylation occurs in a broad range of cancers including endometrioid ovarian and endometrial cancers. Inhibition of the PI3K/Akt/mTOR pathway represents an attractive therapeutic approach (42).
WNT Pathway
β-catenin (CTNNB1) is involved along with cadherins in cell-cell adhesion junctions and may play a role in inhibition of excessive growth when cells come in contact with each other. β-catenin also may be translocated to the nucleus and play a role in regulating transcription of genes involved in embryonic development, cell differentiation, and cell polarity (Fig. 2.6). β-catenin activity is regulated by the WNT pathway (31). The WNT family of genes encodes secreted peptides that interact with Frizzled family cell surface receptors. Frizzled activates the intracellular Dishevelled protein resulting in an increase in the amount of β-catenin translocated to the nucleus. Dishevelled accomplishes this by inhibiting a complex of proteins that includes axin, GSK-3β, and APC that normally promote proteolytic degradation of β-catenin. This allows β-catenin to enter the nucleus and interact with TCF/LEF family transcription factors to promote gene expression.
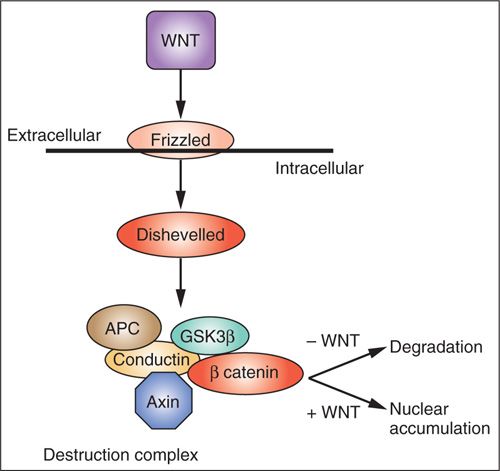
FIGURE 2.6. Wingless (WNT)/β-catenin signaling. WNT extracellular ligands bind Frizzled receptors and regulate the phosphorylation status of axin. Axin functions as part of the destruction complex that regulates the stability of β-catenin, a transcriptional regulator.
Genes encoding WNT signaling inhibitors are often downregulated during carcinogenesis and driver mutations in several of these genes (APC, Axin, GSK-3β, β-catenin) occur frequently in human cancers, including endometrial cancers. Germline mutations in the APC gene are responsible for familial adenomatous polyposis of the colon.
Nuclear Oncogenes
If proliferation is to occur in response to signals generated in the cell membrane and cytoplasm, these events must lead to activation of nuclear transcription factors and other gene products responsible for stimulating DNA replication and cell division (Fig. 2.5A). Expression of several genes that encode nuclear proteins increases dramatically within minutes of treatment of cells with peptide growth factors. Once induced, the products of these genes bind to specific DNA regulatory elements and induce transcription of genes involved in DNA synthesis and cell division. Examples include the FOS and JUN genes, which dimerize to form the AP1 transcription complex. When inappropriately overexpressed these transcription factors can act as oncogenes. The MYC family of nuclear transcription factors is often involved in the development of human cancers, including some ovarian cancers, due to amplification/overexpression. Many of the nuclear regulatory genes such as MYC that control proliferation also impact the threshold for apoptosis. Thus, there is overlap in the molecular pathways that regulate the opposing processes of proliferation and apoptosis.
Genes involved in chromatin remodeling also that have been implicated as oncogenes (e.g., ARID1A). Finally, genes that positively regulate cell cycle progression may be amplified and/or overexpressed in some cancers leading to unrestrained proliferation (e.g., cyclin D1 [CCND1] and cyclin E1 [CCNE1]).
Nuclear Tumor-Suppressor Genes
The retinoblastoma gene (RB) was the first tumor-suppressor gene discovered and is frequently mutated in human cancers (43,44). It was named based on its discovery in the context of a rare hereditary cancer syndrome, as have many other tumor-suppressor genes. The RB gene plays a key role in regulation of cell cycle progression. In the G1 phase of the cell cycle RB protein binds to the E2F family of transcription factors and prevents it from activating transcription of other genes involved in DNA replication and cell cycle progression. This serves as an important restriction point that can protect genomic integrity if DNA damage is present. G1 arrest is maintained by cyclin dependent kinase (cdk) inhibitors such as p16, p21, and p27 that prevent phosphorylation of RB (45). When RB is phosphorylated by cyclin/cdk complexes, E2F is released and stimulates entry into the DNA synthesis phase of the cell cycle. Other cyclins and cyclin dependent kinases are involved in progression from G2 to mitosis. Mutations in the RB gene have been noted primarily in retinoblastomas and sarcomas, but less frequently in other types of cancers. By maintaining G1 arrest, the cdk inhibitors p16, p21, p27, and others act as tumor-suppressor genes. Loss of p16 (CDKN2A) tumor-suppressor function due to genomic deletion or promoter methylation occurs in some cancers. Likewise, loss of p21 and p27 has been noted in some cancers as well.
Mutation of the TP53 tumor-suppressor gene is the most frequent genetic event described thus far in human cancers (46,47). The TP53 gene encodes a 393 amino acid protein that plays a central role in the regulation of both proliferation and apoptosis. In normal cells, p53 protein resides in the nucleus and exerts its tumor-suppressor activity by binding to transcriptional regulatory elements of genes, such as the cdk inhibitor p21, that act to arrest cells in G1. The MDM2 gene product degrades p53 protein when appropriate, whereas p14ARF down-regulates MDM2 when up-regulation of p53 is needed to initiate cell cycle arrest.
Many cancers have missense mutations in one copy of the TP53 gene that result in substitution of a single amino acid in exons 5 through 8, which encode the DNA binding domains that are involved in regulating transcription (Fig. 2.7). Although these mutant TP53 genes encode full-length proteins, they are unable to bind to DNA and regulate transcription of other genes. Mutation of one copy of the TP53 gene often is accompanied by deletion of the other copy, leaving the cancer cell with only mutant p53 protein. If the cancer cell retains one normal copy of the TP53 gene, mutant p53 protein can complex with wild-type p53 protein and prevent it from oligimerizing and interacting with DNA. Because inactivation of both TP53 alleles is not required for loss of p53 function, mutant p53 is said to act in a “dominant negative” fashion. While normal cells have low levels of p53 protein because it is rapidly degraded, missense mutations encode protein products that are resistant to degradation. The resultant overaccumulation of mutant p53 protein in the nucleus can be detected immunohistochemically. A smaller fraction of cancers have mutations in the TP53 gene that encode truncated protein products. In these cases, loss of the other allele occurs as the second event as is seen with other tumor-suppressor genes.
Beyond simply inhibiting proliferation, normal p53 is thought to play a role in preventing cancer by stimulating apoptosis of cells that have undergone excessive genetic damage. In this regard, p53 has been described as the “guardian of the genome” because it delays entry into S phase until the genome has been cleansed of mutations. If DNA repair is inadequate, p53 may initiate apoptosis, thereby eliminating cells with genetic damage (Junttila and Evan, 2009).
TGF-β Pathway
The transforming growth factor-beta (TGF-β) family of growth factors inhibits proliferation (48). It is thought that TGF-β causes G1 arrest by inducing expression of cyclin dependent kinase inhibitors such as p27. Three closely related forms of TGF-β have been discovered that are encoded by separate genes (TGF-β1, TGF-β2, TGF-β3). TGF-β is secreted in an inactive form bound to a portion of its precursor molecule from which it must be cleaved to release biologically active TGF-β. Active TGF-β interacts with type I and type II cell surface TGF-β receptors and initiates serine/threonine kinase activity (49). Prominent intracellular targets include a class of molecules called SMADs that translocate to the nucleus upon TGF-β receptor mediated phosphorylation and act as transcriptional regulator. In the nucleus, the SMAD complex interacts with other DNA-binding transcription factors and co-factors to regulate the transcription of TGF-β target genes to initiate growth arrest. Typical events include the upregulation of cyclin-dependent kinase inhibitors (p16, p15, p21, and p27) and translation-inhibitory protein 4eBP1 as well as the down regulation of MYC. In addition to growth arrest, TGF-β also limits tumor progression through the induction of apoptosis.
MicroRNA
MicroRNAs (miRNA) consist of single RNA strands of about 21 to 23 nucleotides that do not encode proteins (Fig. 2.8). They bind to messenger RNAs that contain complementary sequences and can block protein translation. In addition to regulation of normal biological processes, dysregulation of miRNA expression appears to play a role in the development and behavior of human cancers (50).
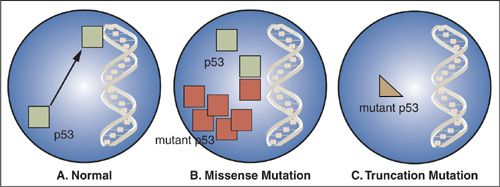
FIGURE 2.7. Inactivation of the p53 gene. A: Normal p53 protein binds to transcriptional regulatory elements in DNA. B: p53 missense mutations encode proteins that no longer bind to DNA and the mutant protein complexes with and inactivates any remaining normal p53 in the nucleus. C: p53 mutations that encode truncated protein products result in proteins that no longer bind to DNA, and these mutations usually are accompanied by deletion of the wild-type p53 allele.
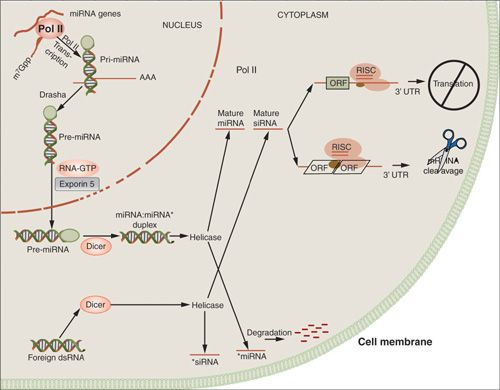
FIGURE 2.8. MicroRNA pathways.
MiRNA production begins in the nucleus from several kilobase-long primary miRNAs (pri-miRNA). These pri-miRNAs undergo cleavage to produce short hairpin-shaped intermediates known as precursor miRNAs (pre-miRNA), which are approximately 70 nucleotides in length.
Drosha, a RNAse III enzyme, along with the DiGeorge syndrome critical region gene 8 (DGCR8), processes the precursors prior to translocation to the cytoplasm via Exportin-5. Subsequently, pre-miRNAs are cleaved by Dicer, also an RNAse III enzyme, resulting in mature miRNAs. SiRNA, an approximately 21-nucleotide single-stranded sequence, is derived within the cytoplasm and does not require processing by the Drosha-DGCR8 complex. Mature miRNAs and siRNAs are then activated through the RNA-induced silencing complex, which produces host mRNA degradation and/or translational repression upon binding to the target. High Dicer and Drosha expression have been shown to be associated with increased median survival in ovarian cancer (>11 years, vs. 2.7 years) (51).
Exogenous constructs of either siRNAs or short hairpin RNAs (shRNA) can also be introduced into cells through transfection. ShRNAs are introduced through viral vectors to produce stable gene silencing, whereas siRNA transfections are more transient. RNAi technology has been used as a research tool to study the effect of turning off various genes and also potentially has therapeutic applications.
Energy Metabolism
Cancer cells uptake increased amounts of glucose to satisfy their metabolic demands (52), and this is the basis of fluorodeoxyglucose (FDG)-PET imaging. Normal tissues generate energy using mitochondrial oxidative phosphorylation and only switch to breaking down glucose to derive energy in the absence of oxygen, which leads to the accumulation of lactate. In contrast, glycolysis of glucose to lactate occurs in cancers even in the presence of oxygen, a phenomenon called ‘aerobic glycolysis.’ This is referred to as the Warburg effect, in honor of its discoverer (53). Because cancers often outgrow their blood supply and become hypoxic, the ability to survive using aerobic glycolysis instead of oxidative phosphorylation may be selected for during malignant transformation. Lactate production by cancers may also promote invasion and metastasis by acidifying the microenvironment. Aerobic glycolysis also may serve the increased metabolic requirement for carbon atoms to produce the macromolecules needed to build new cancer cells. If glucose is completely broken down to carbon dioxide via the citric acid cycle, these carbon building blocks are lost.
The hypoxia-inducible transcription factor-1α (HIF-1α) plays an important role in the cellular response to decreased oxygen in the local environment. When oxygen is absent, HIF-1α accumulates and promotes transcription of pro-angiogenesis genes as well as those involved in glucose transport and glycolysis. HIF-1α accumulation may be a consequence of loss of the VHL tumor suppressor gene, providing a link between the loss of a tumor suppressor and the altered metabolic phenotype of malignant cells. Another link between glucose metabolism and cancer is the finding that isocitrate dehydrogenase (IDH), an enzyme involved in glycolysis, is frequently mutated in glioblastomas. The common amino acid changing IDH mutations result in increased production of 2-hydroxyglutarate, which accumulates to high levels and plays a role in the development of these cancers. Activation of the PIK and RAS pathways also regulate glucose uptake and metabolism.
Differences in metabolism between normal and malignant cells represent an appealing therapeutic target. In this regard, there is a suggestion that the diabetic drug metformin may have efficacy in the treatment and prevention of cancer. This appears to be independent of blood glucose level, and the exact mechanism is unclear.
DNA REPAIR
It has been estimated that thousands of mutations occur in humans on a daily basis (54). Cells in many organs such as the skin, gastrointestinal tract, and respiratory tract that are exposed most directly to the environment constantly undergo renewal with shedding of differentiated cells that may contain mutations. Mammalian cells also have highly evolved and complex DNA repair systems to maintain the integrity of the genome. A series of cell cycle checkpoints exist that allow the opportunity to pause for successful DNA repair, or alternatively for cell death if repair cannot be accomplished. DNA damage checkpoints occur at the boundaries between G1/S and G2/M and during S phase and mitotic spindle assembly. These checkpoints serve to protect against genetic damage that can lead to malignant transformation being fixed in the genome.
There are several repair mechanisms that operate on specific types of DNA damage during these checkpoints (55,56), including mismatch repair (MMR), nucleotide excision repair (NER), base-excision repair (BER), homologous recombination (HR) repair and nonhomologous end joining (NHEJ). Loss of DNA repair activity increases the likelihood of mutations being fixed in the genome and this is a hallmark of many cancers.
DNA Mismatch Repair (MMR)
MMR excises nucleotides that are incorrectly paired with the correct nucleotide on the opposite DNA strand (55). It involves recognition of a base pair mismatch, recruitment of repair enzymes, excision of the incorrect sequence and re-synthesis by DNA polymerase using the parental strand as a template (Fig. 2.9). The recognition of small loops generated by insertion or deletion of nucleotides, as well as single base mismatches is primarily accomplished by a complex called MUTS, which is a heterodimer of MSH2 and MSH6. MLH1 and PMS2 are recruited to the site to initiate the subsequent steps of repair, including excision, DNA synthesis, and ligation.
Loss of mismatch repair leads to a “mutator phenotype” in which there is accumulation of genetic mutations throughout the genome, particularly in repetitive DNA sequences called microsatellites. Examples of microsatellite sequences include mono (AAAA), di (CACACACA), and tri (CAGCAGCAGCAG) nucleotide repeats. Replication errors in these repetitive sequences are common and their inefficient repair leads to the propensity to accumulate mutations, and this is referred to as microsatellite instability (MSI). Some microsatellite sequences are in noncoding areas of the genome while others are within genes. It is thought that accumulation of mutations in microsatellite sequences of tumor suppressor genes may inactivate them and accelerate the process of malignant transformation.
About 3% of endometrial cancers arise due to inherited mutations in MMR genes in the context of Lynch syndrome (57,58). Most cases are due to alterations in MSH2 and MLH1, but MSH6 and PMS1, PMS2, and MSH3 mutations also occur. Lynch syndrome is covered in depth in Chapter 3. More frequently, the MLH1 MMR gene is inactivated due to promoter methylation in sporadic endometrioid cancers of the endometrium leading to microsatellite instability.
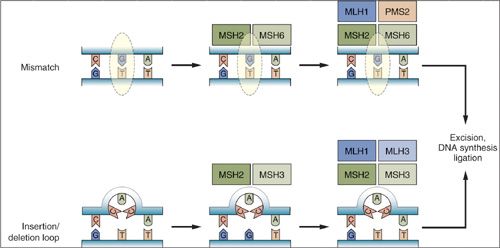
FIGURE 2.9. Mismatch repair pathways. Mispaired bases due to errors in DNA replication or other causes are recognized by the mismatch repair machinery. The initial step involves recognition of simple mismatches by MSH2 and MSH6 (upper panel), or recognition of insertion/deletion loops by MSH2 and MSH3 (lower panel). Subsequent steps involve recruitment of MLH1 and PMS2 to mismatch sites, or MLH1 and MLH3 to insertion/deletion loop sites. This is followed by excision of the respective lesions, DNA synthesis, and ligation to complete the repair.
Nucleotide Excision Repair/Base Excision Repair
The mismatch repair pathway functions primarily in the recognition and repair of replication errors, while the NER and base excision repair (BER) pathways respond to damage caused by DNA damaging agents. NER is an important DNA repair mechanism, as evidenced by the severe human diseases, including Xeroderma pigmentosum, that result from hereditary defects of NER proteins. The NER genes recognize and repair bulky DNA damage caused by environmental carcinogens, ultraviolet light and chemotherapeutic agents such as platinum compounds (Fig. 2.10). Defects in NER proteins predispose to various types of cancer, including skin cancer in Xeroderma pigmentosum (59).
There are two NER pathways: one global pathway involved in scanning the entire genome and another that detects lesions that interfere with elongating RNA polymerases. The proteins required for NER assemble in an ordered stepwise fashion at sites of base damage. This assembly generates a large multiprotein complex namely the “repairosome.” This repair complex can nick the DNA at precise distances on either side of the base damage and these gaps are repaired using the opposite normal DNA strand as a template. There are several proteins involved in NER process including ERCC1, RPA, RAD23A, RAD23B XPA, XPB, XPC, XPD, XPE, XPF, XPG, CSA, and CSB. BRCA1 may also play a role in NER. ERCCI expression may be a marker of cisplatin resistance in cervical cancer, with low expression predicting superior 5-year disease free survival (60).
BER is closely related to NER in that both repair lesions in DNA bases (Fig. 2.10). BER involves repair of small lesions due to chemicals and X-rays that do not distort the DNA helix. Single bases in DNA can be damaged by several mechanisms, the most common being deamination, oxidation, and alkylation. BER is initiated by DNA glycosylases that recognize a single or small set of altered or inappropriate bases. BER removes damaged bases that could otherwise cause mutations by mispairing or lead to breaks in DNA during replication. The BER system excises the inappropriate base from the genome as a free base, leaving a site of base loss in the DNA. These sites are further repaired and reconstructed by a series of biochemical events.
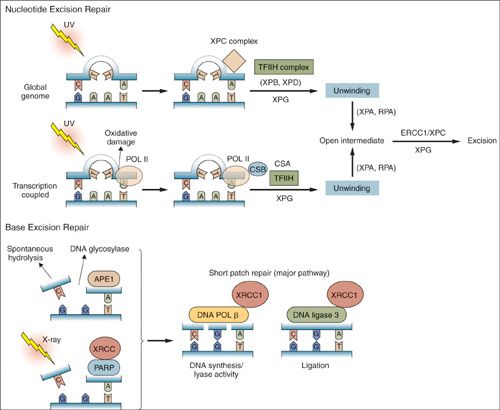
FIGURE 2.10. Nucleotide excision repair and base excision repair. Nucleotide excision repair is activated in response to bulky lesions that are generated, for example, by UV irradiation (upper panels). Global genome repair involves proteins identified by complementation groups in patients with xeroderma pigmentosa (XP proteins). Initial recognition of lesions occurs by a complex containing xeroderma pigmentosa C. Transcription coupled repair also involves proteins identified by mutation in Cockayne syndrome (CS proteins), and occurs when RNA polymerase II stalls at the site of lesions. Stalled RNA polymerase II recruits Cockayne syndrome B to the site of damage. Subsequently, DNA is locally unwound around the injured site by a TFIIH complex containing XPB and XPD. This process also involves XPG, CSA, and other proteins for TCR. Once unwound, XPA and replication protein A contribute to stabilization of an open intermediate and recruitment of the ERCC1 and XPF endonucleases that excise the lesion. Subsequent steps involve DNA synthesis and ligation to complete the repair. In BER (lower panels), abasic sites generated by spontaneous hydrolysis, action of DNA glycosylases, or X-ray–induced single-strand breaks are recognized by the APE1 endonuclease, as well as PARP and XRCC1. Subsequent repair is influenced by PARP-mediated ADP ribosylation of histones and other proteins, while XRCC1 serves as a scaffold for recruitment of DNA polymerase β and DNA ligase 3. These latter enzymes catalyze nucleotide reinsertion and ligation into the injured strand as part of the shor t patch repair pathway (major BER pathway).
Deletion of BER genes increases mutation rates and contributes to the development of various cancers. In this regard, somatic mutations in the DNA polymerase Pol β have been found in some human cancers (61
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


