Molecular Genetics
Charles P. Venditti
Haig H. Kazazian Jr.
The golden age of biology and medical science began around 1945, and can be traced to the work, both basic and applied, of many investigators. These investigators forged the foundation of knowledge that is rapidly revolutionizing modern medicine. The primary event that catalyzed the revolution was the discovery of restriction endonucleases, the bacterial enzymes that cut DNA at precise recognition sites specific for each enzyme. This discovery, along with that of another bacterial enzyme that ligates two pieces of DNA together, led to the ability to produce recombinant DNA—DNA fragments made up of segments from different species. This ability rapidly led to the cloning of human DNA in vectors known to replicate in bacteria, to the production of libraries of DNA fragments, and to the era of recombinant DNA medicine.
In this chapter, the basic principles of gene structure and the basis of recombinant DNA technology are discussed briefly as a prelude to genomics and the Human Genome Project. An outline of what is known about normal variations in DNA follows. The chapter ends with a discussion of mutations and how the knowledge of specific sites of mutation has helped us learn which nucleotide sequences are important in gene expression and how to diagnose the genetic diseases they produce. Also included in this section is a discussion of nonclassical forms of mutation and some unusual modes of inheritance.
GENE STRUCTURE
Genetic material consists of double-helical DNA, with each strand composed of the deoxyribonucleotides, A, G, T, C (adenylic acid, guanylic acid, thymidylic acid, and cytidylic acid, respectively) and a sugar phosphate backbone (Fig. 10.1). This backbone runs along the outside of the helix; the base components of the nucleotides face into the interior of the helix. In that central portion, the critical hydrogen bonds between A of one strand and its complement T of the other, or G of one strand and its complement C of the other, are formed. The human genome contains about 3 billion of these base pairs. The base-pairing rules (A with T and G with C) are critical in information transfer, both during DNA replication and in the transcription of the DNA code into RNA. Each nucleotide is linked to its neighbor via a sugar–phosphate linkage involving the 3′ carbon of one sugar attached to a phosphate group. That phosphate is linked in turn to the 5′ carbon of the next nucleotide. Thus, the nucleotide linkages are referred to as 3′ to 5′. This linkage gives a DNA (or RNA) strand its polarity or directionality. This polarity determines the direction in which the DNA is synthesized during replication and read during the transcription of DNA into RNA. Thus, a DNA sequence 3′ to 5′ is decoded during transcription in a 5′ to 3′ direction in RNA.
The great majority of mammalian genes coding for a protein product have split coding regions; that is, the coding regions are discontinuous. The beta-globin gene of adult hemoglobin has three coding regions, termed exons, and two intervening sequences (IVS), called introns. The coding regions are divided between the codons for amino acids 30 and 3l of the l46 amino-acid chain and the codons for amino acids l04 and l05. Although only about 450 nucleotides are necessary to encode the protein (3 nucleotides/amino acid), because of the introns, the gene contains roughly 1,500 nucleotides. Yet, this is a very simple gene. Many genes now have been described with 10 or more introns (the gene for the pro-alpha 1 collagen chain has more than 50 introns), and some genes have a total size of more than 200 kilobases (kb) or 200 thousand base pairs. In fact, dystrophin, the gene affected in Duchenne muscular dystrophy, has over 2 million base pairs. Some large introns contain entire genes within their boundaries. On the other hand, certain genes, such as those encoding histones and interferons, are small and do not contain introns. These genes are unexplained exceptions to the split gene rule.
EXPRESSION OF GENETIC MATERIAL
The mechanisms of expression for protein-coding genes are exemplified by the globins (Fig. 10.2). The entire gene, including the two introns, is transcribed into a precursor RNA. This RNA is immediately modified at both its ends (called 5′ and 3′ ends). The modification at the 5′ end is an addition of a methylated G in an unusual triphosphate linkage. This modification, which is specific for messenger RNA, is called
5′ capping. The modification at the 3′ end is the addition of about l50 A residues (the poly A tail). The 5′ cap is thought to be important in the translation of the RNA into protein, whereas the 3′ poly A tail may have a role in RNA stability. Next, the introns are precisely spliced out of the RNA, thereby approximating the coding region sequences. The precise mechanism, and the enzymes involved in RNA splicing, is now well understood.
5′ capping. The modification at the 3′ end is the addition of about l50 A residues (the poly A tail). The 5′ cap is thought to be important in the translation of the RNA into protein, whereas the 3′ poly A tail may have a role in RNA stability. Next, the introns are precisely spliced out of the RNA, thereby approximating the coding region sequences. The precise mechanism, and the enzymes involved in RNA splicing, is now well understood.
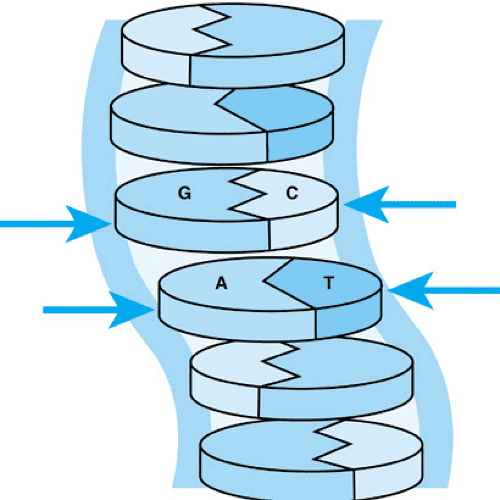 FIGURE 10.1. Base pairing in DNA. As shown, G of one strand pairs by hydrogen bonding with C of the other strand. Likewise, A pairs with T. |
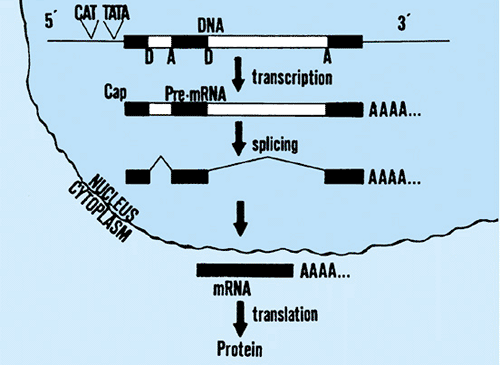 FIGURE 10.2. Gene expression. The dark black bars correspond to exons, the unshaded regions between the D and A represent introns. Steps include (a) transcription of precursor RNA, (b) cap and poly A addition to the ends of the RNA, (c) splicing out of intron sequences, and (d) translation of the mature messenger RNA into protein. CAT and TATA in front of the gene refer to sequences important in transcription, whereas D and A refer to donor and acceptor splice junctions important in RNA splicing. |
RNA splicing also is important in transporting the RNA from the nucleus to the cytoplasm, where it will be translated into protein; nucleotide sequences near the exon–intron junctions are important in normal splicing. Translation then occurs on the fully processed messenger RNA (mRNA) in the cytoplasm to produce the peptide chain, which may need post-translational modification, such as glycosylation or farnysylation, and/or subcellular targeting for proper functioning. Mutations that interfere with any of these steps have the potential to cause human disease.
The genetic code, as found in the mRNA, is critical to translation. There are 64 possible combinations in a code composed of three nucleotides. One combination, AUG (note that U is substituted for T in RNA) is the initiation codon and always places methionine as the initial amino acid in a protein. Methionine usually is not the first amino acid in the finished protein because it often is removed when the nascent protein is in the early stages of production. The code also contains three terminator codons, UAG, UAA, and UGA. These codons cannot be decoded into an amino acid and, therefore, protein synthesis terminates whenever one of these triplets is present in the reading frame. A mutation in the coding region of a gene that produces a terminator codon is called a nonsense mutation. A nucleotide change that produces a mutation by changing the sequence of an amino acid is called a missense mutation. The remaining 60 codons can be decoded so that an amino acid is inserted into the growing or nascent polypeptide chain. The degeneracy of the genetic code allows for six different codons for leucine and serine, and four codons for many other of the total of 20 amino acids. This degeneracy permits some variation in the coding DNA sequence, without necessarily producing a change in the amino acid encoded. The relationship between the nucleotide triplet in DNA, its complement in mRNA, and the amino acid designated by that triplet is demonstrated for the sixth codon of the beta-globin chain (Fig. 10.3). CTC in DNA is decoded as GAG in mRNA and as glutamate in the protein. A single nucleotide substitution (T to A) causes a missense mutation that leads to sickle cell anemia, as discussed later.
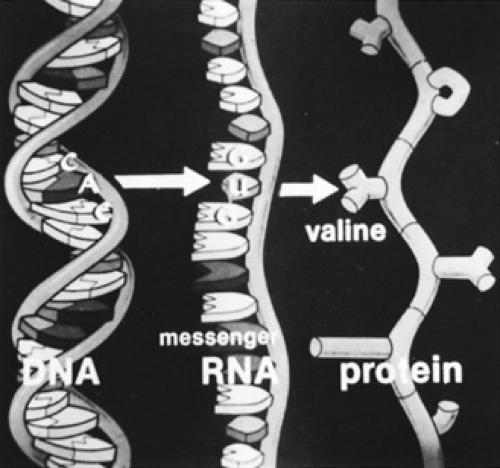 FIGURE 10.3. Relationship between the triplet code in the strand of DNA that is transcribed, the codon in messenger RNA, and the specific amino acid inserted in the polypeptide chain. For example, CAC in the DNA is transcribed into GUG in RNA and the amino acid inserted is valine. Shown here is the mutant sequence in the beta-globin gene for the betas or sickle hemoglobin chain. The sequence of normal betaA-globin DNA for the strand shown is CTC. |
NORMAL VARIATION IN DNA
Any discussion of mutations and their consequences requires a brief introduction to normal variation. Much has been learned about normal variation at the protein or enzyme level. Scientists are learning that variations in the DNA are even more extensive than we imagined from protein data. A number of different types of variation have been found. Originally, common normal variation in DNA was termed DNA polymorphism, and it could be detected in the laboratory as a restriction fragment length polymorphism (RFLP). This variation usually is the result of single nucleotide substitutions and is detected if the variation affects a restriction endonuclease site. The insertion or deletion of a large number of nucleotides also can be detected as an RFLP. One RFLP that affects a site that is cleaved by the restriction enzyme HincII near the beta-globin gene is shown (Fig. 10.4). When genomic DNA is digested with HincII, electrophoresed, and hybridized with a radioactive probe containing the beta-globin gene, chromosomes that contain this site yield a 3.7-kb fragment, and those that lack the site demonstrate an 8-kb fragment. Because this polymorphism is very frequent, nearly 50% of individuals in the population are heterozygous and demonstrate both fragments by this analysis.
Southern blotting (Box 10.1) is a technique used to find a number of DNA polymorphisms in a gene cluster and in and around a large number of other genes. Thus, normal variation in DNA is extensive. The 50,000 nucleotides of the paternal beta-globin gene cluster contain l00 or more nucleotide differences from the 50,000 maternal nucleotides of this cluster. In total, the haploid DNA derived from one parent contains 3 to 10 million differences (single nucleotide substitutions) from the genetic material derived from one’s other parent.
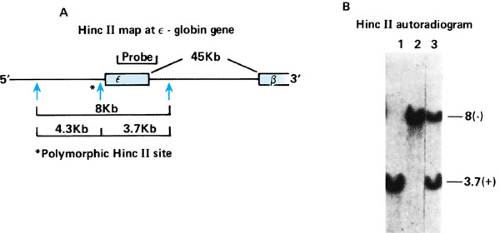 FIGURE 10.4. A polymorphism of a restriction endonuclease site near the epsilon-globin gene. A: Near many normal epsilon-globin genes is a HincII site, but this site is often missing in normal individuals. When the site is present, the 8-kb fragment is cut into 3.7- and 4.3-kb fragments. B: The 3.7-kb fragment is readily detected by use of a DNA probe containing the epsilon gene. When the site is absent, the 8-kb fragment is not cleaved. The presence or absence of this HincII site can be discovered by Southern blotting (see Fig. 10.5). |
BOX 10.1 Molecular Techniques: Southern Blotting
Single gene fragments are detected by a technique called Southern blotting. Southern blotting is named for E. M. Southern, inventor of the method, and it can be used to detect single copy genes in as little as 3.5 micrograms of DNA. [See section, General Methods of DNA Analysis, for a discussion of the polymerase chain reaction (PCR), which can detect single genes in 100 nanograms of DNA routinely, and in 10 picograms of DNA in research labs. Ten picograms is the amount of DNA in a single human sperm.] Most Southern blot analyses are carried out on DNA isolated from the leukocytes of peripheral blood, from which one can isolate about 50 to l00 μg/mL of blood (Fig. 1). Usually 5 μg of genomic DNA of an individual is digested with a specific restriction enzyme, and the digested DNA is subjected to electrophoresis in an agarose gel that separates DNA on the basis of size. The DNA is transferred to a nitrocellulose paper and hybridized to a radioactive cloned DNA fragment of interest (the probe). After hybridization with the probe, the filter is washed to remove nonspecific radioactivity, and the washed filter is placed in contact with an x-ray film. After a day or two, the film is removed and developed to demonstrate the bands of interest.
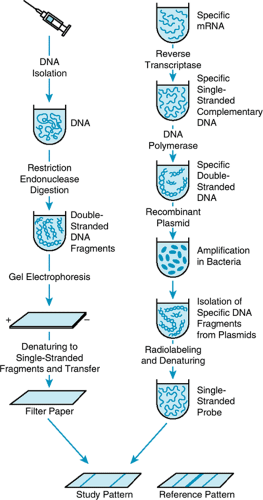 BOX 10.1. FIGURE 1. Southern Blotting, or Restriction Endonuclease Analysis. The steps in this procedure are shown, beginning with isolation of DNA from leukocytes to discovery of hybridizing fragments (bands) in that DNA with a specific radioactive probe. |
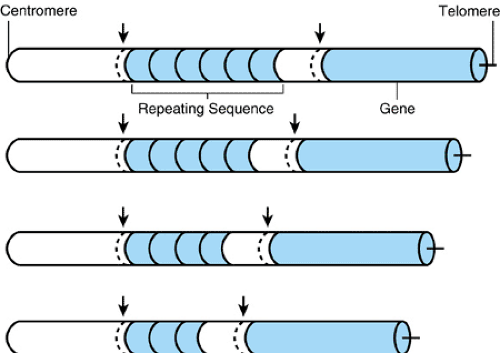 FIGURE 10.5. A VNTR (variable number of tandem repeats) polymorphism adjacent to a gene. A 30-nucleotide sequence has three repeats, four repeats, five repeats, or six repeats in tandem in different chromosomal homologues. The length variation in this region of the genome can be easily detected by Southern blot analysis or the polymerase chain reaction. In an SSR (simple sequence repeat) polymorphism, the polymorphic repeat sequence would contain two to four nucleotides (e.g., the dinucleotide CA), repeated ten to 30 times. |
Even more important forms of DNA polymorphisms are variable number of tandem repeats (VNTRs) and simple sequence repeats (SSRs). VNTRs are repeats of 15 to 50 nucleotides, and the repeat number at a locus may vary widely (Fig. 10.5). SSRs are variations in the number of repeats of very short sequences, usually two to four nucleotides. Often, a VNTR locus may contain many possible alleles, perhaps 100 or more, if a population group is studied. SSR loci may contain 5 to 10 alleles. The variation in VNTRs and SSRs contrasts with that of most common RFLPs, which are dimorphisms. Because of the large number of alleles at many VNTR loci, a particular genotype at four or five such loci may be present in only one person in several million individuals in the population. This normal DNA variation has had substantial practical value in the forensic investigation of criminal cases. DNA analysis has played a major role in the conviction or exclusion of many individuals accused of rape or other crimes.
DNA polymorphisms also have been used as markers to trace the inheritance of particular regions of the genome in families. When a large number of such markers are studied in families affected with single-gene diseases, such as Huntington disease, it has been possible to find a single marker that is co-inherited with the disease gene in the affected families. After this marker is mapped to some particular chromosome segment, the disease gene is ipso facto mapped to the same location. In this way, the Huntington disease (HD) locus was mapped to the short arm of chromosome 4, the neurofibromatosis locus (NF-1) to the centromeric region of chromosome 17, and the cystic fibrosis locus (CF) to the long arm of chromosome 7, to name a few successful examples of this approach.
The genes responsible for these diseases have been isolated by this method of finding the gene’s location in the genome before knowing the function of the encoded product. One of the great promises of the Human Genome Project is that this type of reverse genetic analysis, called positional cloning, will be very easy to execute. Once a gene is mapped to a region of the chromosome, and the DNA sequence of that region is known, finding genes in that region can be done quickly using a combination of computational and experimental methods. For example, if a metabolic disorder known to be associated with an electron transport chain defect is localized to a specific chromosomal region, a researcher might examine the DNA sequence in that region carefully for gene products that are predicted to have a role in energy metabolism and then directly sequence those genes in the DNA from affected patients to determine causation. This sequence of events has been performed a number of times with great rapidity since the genome sequence has become available.
GENOME ORGANIZATION AND THE HUMAN GENOME PROJECT
In the late 1980s, the idea of determining the exact DNA sequence of the entire human genome was discussed. By the late 1990s, the project was well underway. Two remarkable reports appeared in February 2001, describing the sequencing and analysis of most of the human genome, completed several years earlier than anticipated. The availability of this sequence or blueprint for life is certain to revolutionize the impact of genetics in pediatrics and in medicine in general. For that reason, we review selected aspects of the human genome sequencing project. The strategies for mapping and sequencing the human genome are described in Box 10.2, Figure 1.
Human Genome Project: Selected Findings
The analysis of the DNA sequence of the human genome has provided fundamental insights into evolution, chromosome structure and recombination, the origin and organization of genes, and DNA and protein polymorphisms. It also has identified the set of protein coding genes.
The human genome is largely composed of interspersed repeat elements of various classes. Long interspersed nuclear elements (LINEs); short interspersed nuclear repeats (SINEs), including the Alu elements); long terminal repeats (LTRs); and retrotransposons and DNA transposon copies comprise 21%, 13%, 8%, and 3%, respectively, of the genome sequence and total over 3 million copies—three orders of magnitude greater than the largest multigene family. Retrotransposons are copied into RNA, reverse transcribed into complementary DNA (cDNA), and then inserted into the genome at a new location. Transposons are DNA sequences that are directly “cut and pasted” into another genomic site. Alu sequences are the most prevalent constituent of the genome. Alu sequences contain an AluI restriction endonuclease site, are about 300 nucleotides in length and, when compared with one another, are roughly 95% homologous.
Transposable elements, once considered “junk” DNA, are thought be a driving force in genome evolution, because they can be associated with recombination, pseudogene formation, and exon shuffling. It is now known that some copies of LINE elements are capable of retrotransposition and have been shown to produce human disease when they are mobilized to a new genomic location (see section, Insertion of Transposable Elements). The occurrence of repeated sequences located in the flanking DNA between functional gene sequences and within introns of genes is a general characteristic of the human genome. It is likely that repetitive DNA is not “junk,” but part of an evolutionary engine that has shaped the human genome.
Single Nucleotide Polymorphisms
As discussed, the individual base sequence at any position in the DNA can be either A, T, G, or C. In certain positions, scattered throughout the genome, are variations at only one such nucleotide in a stretch. These changes are called single nucleotide polymorphisms (SNPs). When the draft sequence of the human genome was analyzed, 1.42 million such changes were detected.
They occur roughly once per 1,000 base pairs. Approximately 60,000 of these polymorphisms were noted to fall in the coding and untranslated regions of genes; some change the amino acid sequence of constituent genes, generating polymorphism at the protein level. Because these changes are detected easily using methods that are amenable to high throughput analysis, it is possible to study SNPs as functional genetic markers.
They occur roughly once per 1,000 base pairs. Approximately 60,000 of these polymorphisms were noted to fall in the coding and untranslated regions of genes; some change the amino acid sequence of constituent genes, generating polymorphism at the protein level. Because these changes are detected easily using methods that are amenable to high throughput analysis, it is possible to study SNPs as functional genetic markers.
BOX 10.2 Molecular Technique: The Human Genome Project
The strategy to map and sequence the human genome relied on an integration of a variety of technologies. A general overview is presented in Figure 1. Genetic and cytogenetic markers were used to define a set of large-insert DNA clones called BACs (bacterial artificial chromosomes) isolated from a library of human DNA. These BACs contained large pieces of human DNA, 100 to 250 kilobase pairs, that could be tracked to the origin and exact location of the specific human chromosome from which they were derived. The BAC clones were then mapped using restriction enzyme digestion and gel electrophoresis to generate “fingerprints” for each clone. Overlapping clones then could be identified with certainty, because they shared a similar fingerprint pattern, thus allowing groups of such clones to be ordered into larger units called contigs. These contigs in turn could be assigned to a specific portion of a chromosome and the map position or location also could be verified using other parallel methods, such as fluorescent in situ hybridization (FISH) and radiation hybrid (RH) mapping. Thus, the starting material in this approach was a physically mapped collection of overlapping large-insert BAC clones.
To generate the raw sequence for the genome analysis, the mapped BAC clones were then uniformly fragmented by shearing or restriction enzyme digestion and re-cloned as small fragments for random sequencing. Robotic technology and computational advances allowed this process, called shotgun sequencing, to be performed on a large scale. One factory-style robotic instrument used in the project was able to process 100,000 sequencing reactions in 12 hours. Considering that, a decade ago, it took one person an entire day to process 10 to 20 similar specimens, the technologic advances required to execute this project can be more fully appreciated. The small overlapping shotgun clone sequences were then assembled by looking for overlap in the DNA sequence output, followed by alignment to generate the entire sequence of the parent BAC clone. A reiteration of this process using DNA sequences from different BACs allowed larger regions to be similarly aligned, eventually connecting all the contiguous large insert clones together.
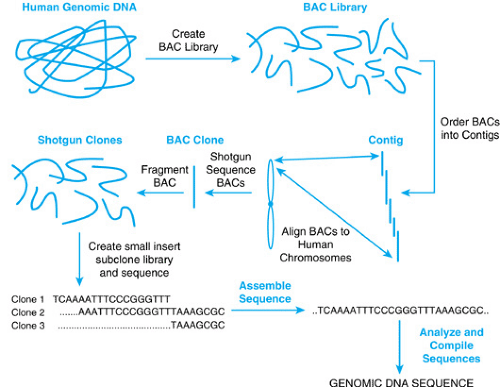 BOX 10.2. FIGURE 2. An overview of the Human Genome Sequencing Strategy. A large insert DNA library was prepared by size fractionating human genomic DNA. The fragments were cloned as bacterial artificial chromosome (BACs) clones, assembled by a variety of methods, including fingerprinting, into contigs. The ordered BACs were then fragmented and recloned as random small fragments which were then sequenced and assembled to produce the DNA sequence of the genome. |
The genomic sequences generated in this fashion then can be further analyzed using computer programs, as well as other resources, in an attempt to annotate the sequence. One important approach has been to try to correlate the sequence of expressed genes with their location and their intron–exon boundaries by analyzing complementary DNA. Complementary DNA (cDNA) is derived from purified messenger RNA that has been isolated from a tissue, such as white blood cells or muscle, and has been copied into DNA using a retroviral enzyme called reverse transcriptase. The resulting cDNA can be transferred into a vector that replicates in bacteria, allowing individual cDNA clones to be isolated and sequenced. Comparisons between the genomic DNA and the sequence of cDNAs from a variety of tissue sources helps define the structure of each gene. Multiple techniques—both computational and experimental—then can be used to define individual genes in the genome. It is important to realize that not all genes can be identified by any one method; therefore, a combination of techniques is required to make robust predictions. Other methods that utilize large-scale analysis of proteins (proteomics) will ultimately be needed to confirm the predictions made from sequence analysis.
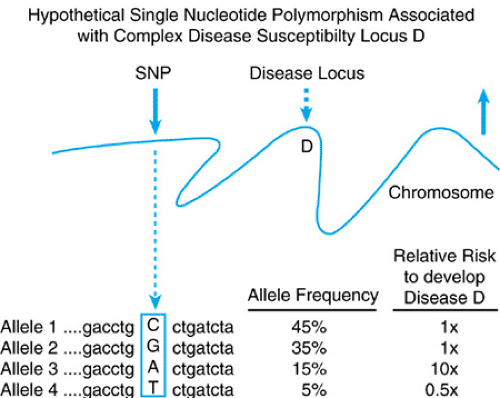 FIGURE 10.6. A hypothetical SNP and its association with a disease susceptibility locus D. This figure depicts a specific DNA change (SNP indicated by the bold arrow) and the association it has with susceptibility locus D. In this example, allele 3 is associated with an increased relative risk and allele 4 a decreased risk. |
Furthermore, because certain combinations of SNPs are preferentially associated together in blocks throughout the genome, they should allow the expedient execution of genetic association studies. Although certain SNP alleles may be associated with increased risks to develop multifactorial diseases, such as diabetes, by virtue of causation or proximity, others may confer protective effects. Figure 10.6 displays what a hypothetical SNP is and how it may be used to make predictions about disease susceptibility. SNP analysis will be important in dissecting the etiology of diseases that exhibit gene–environment interactions and complex inheritance, such as autoimmune, endocrine, behavioral, and developmental disorders. In the future, the simultaneous genetic testing of multiple SNPs might help augment preventive medicine efforts. Several examples of the utility and power of using these genetic markers have appeared; these include their use to identify susceptibility loci for systemic lupus erythematosus, type 2 diabetes, and inflammatory bowel disease.
Gene Organization and Protein Coding Gene Number
The analysis of the human genome sequence has documented that a typical human gene spans ∼20 kilobase pairs and contains seven to eight small exons. One area of controversy and debate had been the predictions of human gene number. Earlier estimates had ranged between 50,000 and 100,000 protein coding genes, despite the fact that kinetic reassociation studies of mRNA suggested otherwise. It now appears that the actual number of genes is between 25,000 and 30,000, and the actual protein-coding fraction of the genome is only about 1% to 2%. Figure 10.7 shows the distribution of various protein families in humans encoded by the human genome.
One major difference between human genes and those from lower organisms appears to be that human protein coding genes have evolved new architectures by adding, deleting, and reordering existing motifs to create new proteins and expand existing families. If one also considers that many genes are alternatively spliced to generate different isoforms, often in a tissue- or developmental-specific fashion, it is possible to envision that the capacity for increased interactions in the human protein set or proteome is much greater than that accounted for by the linear extrapolation of increased gene numbers in humans versus other model organisms, such as Drosophila or Caenorhabditis elegans. In addition to protein encoding genes, large numbers of non-protein coding RNAs, such as tRNAs, rRNAs, and small nuclear RNAs have been characterized with similar interest. It is likely that many small noncoding regulatory RNAs also exist and have yet to be fully annotated and functionally defined. In fact, in a recent study that compared large genomic sequences from human to several vertebrate species, a substantial number of previously unrecognized, conserved noncoding segments were noted. These findings indicate that a large reservoir of poorly understood but functionally important DNA sequences exist in the human genome and intimate that new insights into human genetic disease will evolve from examining these sequences in health and disease.
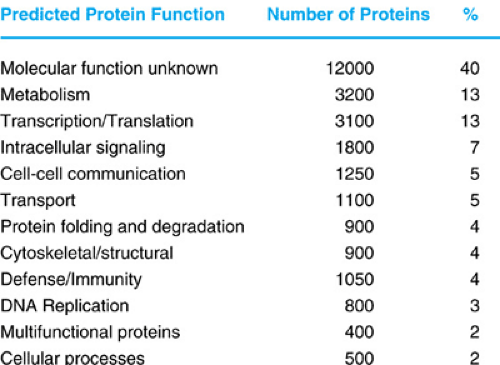 FIGURE 10.7. The function of predicted and known families encoded by the human genome. The approximate number of proteins and percent of the total are indicated. Currently, the largest fraction of genes encode proteins of unknown function.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|