Chapter 486 Molecular and Cellular Biology of Cancer
Genes Involved in Oncogenesis
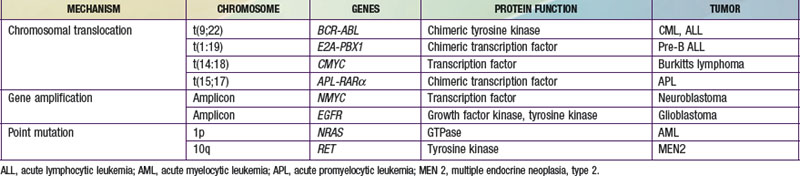
The three main mechanisms by which proto-oncogenes can be activated include amplification, point mutations, and translocation (Table 486-1). MYC, which codes for a protein that regulates transcription, is an example of a proto-oncogene that is activated by amplification. Patients with neuroblastoma in which the MYC gene is amplified 10-300-fold have a poorer outcome. Point mutations can also activate proto-oncogenes. The NRAS proto-oncogene codes for a guanine-nucleotide-binding protein with guanosine triphosphatase activity that is important in signal transduction and is mutated in 25-30% of acute nonmyelogenous leukemias, resulting in a constitutively active protein. The RET protein is a transmembrane tyrosine kinase receptor that is important in signal transduction. A point mutation in the RET gene results in the constitutive activation of a tyrosine kinase, as found in multiple neoplasia syndromes and familial thyroid carcinoma.
Syndromes Predisposing to Cancer
Several syndromes are associated with an increased risk of developing malignancies, which can be characterized by different mechanisms (Table 486-2). One mechanism involves the inactivation of tumor suppressor genes such as RB in familial retinoblastoma. Interestingly, patients with retinoblastoma in which one of the alleles is inactivated throughout all of the patient’s cells are also at a very high risk for developing osteosarcoma. A familial syndrome, Li-Fraumeni syndrome, in which one mutant P53 allele is inherited, has also been described in patients who develop sarcomas, leukemias, and cancers of the breast, bone, lung, and brain. Neurofibromatosis is a condition characterized by the proliferation cells of neural crest origin, leading to neurofibromas. These patients are at a higher risk of developing malignant schwannomas and pheochromocytomas. Neurofibromatosis is often inherited in an autosomal dominant fashion, although 50% of the cases present without a family history and occur secondary to the high rate of spontaneous mutations of the NF1 gene.
Table 486-2 FAMILIAL OR GENETIC SUSCEPTIBILITY TO MALIGNANCY
| DISORDER | TUMOR/CANCER | COMMENT |
|---|---|---|
| CHROMOSOMAL SYNDROMES | ||
| Chromosome 11p-(deletion) with sporadic aniridia | Wilms tumor | Associated with genitourinary anomalies, mental retardation, WT1 gene |
| Chromosomal 13q-(deletion) | Retinoblastoma, sarcoma | Associated with mental retardation, skeletal malformations; autosomal dominant (bilateral) or sporadic new mutations, RB1
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|