Mineral Metabolism in the Newborn
Joseph M. Gertner
SKELETAL DEVELOPMENT IN THE FETUS AND NEONATE
Histogenesis and Organogenesis of the Skeleton
At the end of a 40-week term of gestation, the neonatal skeleton has reached an organization close to that of the adult. The development of the skeleton depends on the integration of events directing cells derived from primitive mesenchyme to produce bone matrix, mineralize that matrix, and then remodel the resulting bone. This entire process is under complex genetic control and can be distorted by disordered formation of fibrous and nonfibrous matrix proteins, faulty migration of bone-forming cells, the failure to recruit the appropriate cells for bone remodeling, and abnormalities of the hormonal and ionic milieu needed to promote mineralization.
The precursors of bone cells begin to form at approximately 5 weeks’ gestation from membranes or rods of mesenchymal cells. Intramembranous bone forms the sides and vault of the skull and the clavicles, whereas endochondral ossification accounts for most of the remaining bones, particularly those of the developing limbs. In both forms of ossification, osteoprogenitor cells condense, mature into alkaline phosphatase–positive osteoblasts, secrete an extracellular ground substance, and begin to mineralize. A network of collagen fibers forms the framework on which the first bony trabecula mineralizes to form the so-called primary spongiosa. Secondary foci of ossification that eventually become epiphyseal centers differentiate from mesenchyme in an analogous manner. Remodeling of the skeleton begins as soon as the primary spongiosa is formed; osteoclasts resorb existing bone, whereas osteoblasts form new bone.
Ionic and Hormonal Effects on the Fetal Skeleton
The mineralization process depends on the controlled delivery of calcium and phosphate to the sites of ossification and on the local effects of the calciotropic hormones (i.e., parathyroid hormone [PTH], calcitriol (1,25-dihydroxyvitamin D), and calcitonin). Calcium and phosphorus are transported across the placenta against a concentration gradient that appears as early as 12 weeks. The quantity transported increases sharply until late in the last trimester, by which time the fetus is accumulating up to 85 mg/kg/day of phosphorus and 150 mg/kg/day of calcium (Fig. 64.1). Placental calcium transport depends on the availability of calcitriol and the PTH-related peptide (PTHrP), but no known intrinsic disorders of the placenta exist that specifically limit calcium and phosphorus accumulation. Mineral deficiency may accompany the general fetal malnutrition characteristic of placental insufficiency. Pathologic consequences can arise from disordered transplacental calcium transport when calcium concentrations in the maternal serum are too high or too low. These disorders are considered in detail in the following section.
PTH is an 84–amino acid peptide secreted by the parathyroid glands that elevates serum ionized calcium by promoting bone resorption. The parathyroid glands are formed from cells of the third and fourth pharyngeal pouches at the sixth to seventh week of gestation and stain positively for PTH by the twelfth week. The parathyroid glands are the principal regulators of extracellular ionized calcium concentration. The chief cells serve both as calcium sensors and effectors of calcium homeostasis. A calcium ion–sensitive G-protein–linked receptor (CaSR) detects ionized calcium concentration in the extracellular fluid and modifies secretion of PTH. Fetal hypoparathyroidism is without effect on the fetus because maternal calcium homeostasis prevails in utero. Intrauterine hyperparathyroidism, on the other hand, can lead to pathologic resorption of the fetal skeleton.
The C cells of the thyroid are derived from ultimobranchial (fifth pharyngeal pouch) tissue and also begin to secrete their 22–amino acid peptide, calcitonin, at an early stage of gestation. Acutely, calcitonin lowers the extracellular ionized calcium concentration by inhibiting bone resorption. However, humans of all ages maintain normal calcium homeostasis and bone turnover in the absence of calcitonin (e.g., in athyreotic states) and in the presence of large excesses of the hormone (medullary carcinoma of the thyroid). The physiologic function of calcitonin, both in utero and in the neonatal period, is thus obscure. It has been suggested that the neonatal hypercalcitoninemia contributes to neonatal hypercalcemia.
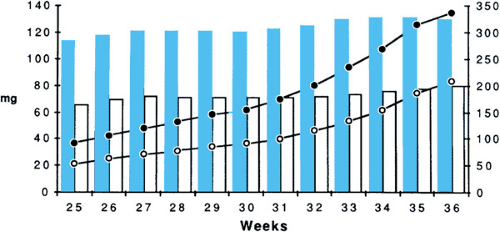 FIGURE 64.1. The intrauterine accumulation of phosphorus and calcium during late pregnancy. White circle, phosphorus (mg/day); black circle, calcium (mg/day). |
A third calciotropic hormone, calcitriol, is a lipid-soluble sterol that, unlike the two peptide hormones, can be transported across the placenta. Although clear evidence shows that both the fetal kidneys and the placenta itself can make calcitriol, we do no know what proportion of the hormone is transported from the maternal circulation as opposed to synthesis in the fetus or placenta. However, It has been established that the precursor sterol, calcidiol (25-hydroxyvitamin D), is transported across the placenta. Its concentration in fetal blood is highly correlated with the maternal level, and it is needed for fetal synthesis of calcitriol. The major biologic action of calcitriol, the promotion of intestinal calcium absorption, is, of course, unnecessary during fetal life. The role of fetal vitamin D metabolites in the process of bone development is unclear, but the existence of congenital rickets as a pathologic entity suggests that vitamin D sterols do exert some direct effect on mineralization.
Another substance, discovered more recently than PTH or vitamin D, that may affect prenatal and perinatal bone mineral physiology is PTHrP, which has come under investigation. This 141–amino acid peptide was first characterized in malignant cells derived from patients with the syndrome of hypercalcemia of malignancy. PTHrP shares a region of homology with PTH, encompassing the 13 N-terminal amino acids, and it shares with PTH the power to bind to the type 1 PTH receptor, activating bone resorption and renal tubular phosphate reabsorption. Messenger RNA for PTHrP and the peptide itself are found in the lactating breast, with considerable quantities secreted into the milk, and in placental tissue. The corresponding PTHrP appears to influence the function of the uteroplacental vasculature and to play a part in transplacental calcium transport, but the importance of this role and the effect, if any, of PTHrP on the fetus and infant from the placenta or breast milk remains unclear.
Perinatal Mineral Homeostasis
After delivery, the fetus must adapt to the sudden withdrawal of an abundant placental supply of calcium and phosphate. Mean fetal calcium levels decrease from approximately 11 mg/dL at birth to a nadir of approximately 8.5 mg/dL in full-term and 7.0 mg/dL in premature infants (Fig. 64.2).
These minimum levels of serum calcium occur 1 to 2 days after delivery and give rise to hormonal adjustments that promote the infant’s metabolic adaptation to postnatal mineral homeostasis. PTH levels appear to increase postnatally, an appropriate response to the decrease in serum calcium. Serum calcitonin levels are said to be high in the perinatal period, but little information is available concerning changes during the first few days of postnatal life.
Mean serum calcidiol and calcitriol concentrations are lower in neonates than in mothers but do not differ between full-term and premature babies. In full-term and premature infants, serum calcitriol increases from birth to day 5 and decreases again by day 30, but in preterm infants, calcitriol levels are higher than in full-term infants on day 30. The role of calcitriol in calcium homeostasis depends largely on the promotion of gastrointestinal calcium absorption, so it follows that the postnatal increase in calcitriol will be less effective in infants whose oral calcium intake is inadequate or who have disordered intestinal function. Disorders of calcium control in the neonate can arise because of failures of the PTH–vitamin D homeostatic system or because that system is overwhelmed by unfavorable circumstances. These disorders are discussed in the sections on Neonatal Hypercalcemia and Neonatal Hypocalcemia.
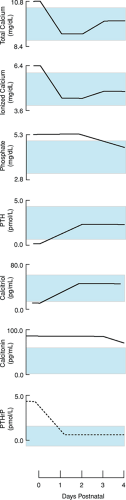 FIGURE 64.2. Changes in biochemical measures in the neonatal period. PTH, parathyroid hormone; PTHrP, parathyroid hormone–related peptide. (Modified from Kovacs CD, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev 1997;18:832.) |
HORMONAL AND METABOLIC DISORDERS
Congenital Rickets
Congenital rickets is now a rare condition but is of theoretical interest in view of the importance of rickets as a component of metabolic bone disease of prematurity. The appearance in the full-term neonate of frayed and cupped epiphyses, with general skeletal rarefaction and sometimes with deformities, was well described in the older literature. The condition accompanies severe maternal vitamin D deficiency and has been attributed to the same defect in the fetus. Some of the appearances are likely caused by fetal hyperparathyroidism, secondary to maternal and fetal hypocalcemia, but some may result directly from deficiency of vitamin D metabolites supplied to the fetus.
Neonatal Hypercalcemia
Williams Syndrome
Congenital facial and cardiovascular abnormalities (supravalvular aortic stenosis and peripheral pulmonary stenosis) and mental retardation are often accompanied by transient infantile hypercalcemia. The hypercalcemia may not be recognized until many weeks of age, but retrospective evidence may be obtained from a history of poor feeding or constipation in the neonatal period.
Epidemiology
Virtually all patients with Williams syndrome are heterozygotes for a fresh mutation (microdeletion at 7q11.23). Familial cases are rare. This microdeletion, detectable by the fluorescent in situ hybridization (FISH), gives rise to a loss of contiguous genes. The loss of the elastin gene probably causes the cardiac and facial abnormalities, whereas the cognitive defect is believed to be caused by mutations in adjacent genes.
Pathophysiology
Despite this genetic evidence, the precise cause of the hypercalcemia, which usually remits between 9 and 18 months of age, remains unknown. Elevations in calcidiol, elevated and subnormal concentrations of calcitriol, and deficient calcitonin release have all been proposed, but little firm evidence exists for any of these theories. The mental retardation is not a consequence of high serum calcium levels.
Fetal and Neonatal Hyperparathyroidism
The fetal parathyroids are operative from the first trimester and respond to reduced extracellular calcium concentration by inducing the resorption of bone. The term neonatal hyperparathyroidism usually refers to primary hyperparathyroidism detected by severe malaise (vomiting, dehydration, constipation) during the first few days of life.
Pathophysiology
The best-defined cause of the condition is parathyroid hyperplasia forming part of a familial syndrome caused by mutations in the gene for the calcium-sensing receptor (CASR). Most such cases are homozygotes for a trait represented by familial hypocalciuric hypercalcemia (FHH) in the heterozygotes.
A rare condition combining skeletal malformations with some of the biochemical manifestations of hyperparathyroidism is Jansen metaphyseal chondrodysplasia. This rare but theoretically interesting skeletal dysplasia was described in 1934 by Jansen. Recently, gain-of-function mutations in the type 1 PTH receptor gene, which is also the receptor for PTHrP, have been shown to cause the condition. These patients are hypercalcemic with suppressed PTH levels. The embryogenesis of the peripheral skeleton seems to require PTHrP, and it may be disruption of the PTHrP system rather than the activation of PTH receptor signaling that leads to chondrodysplasia in affected individuals.
In secondary hyperparathyroidism, the fetal glands are stimulated by fetal hypocalcemia caused by maternal hypocalcemia. These circumstances can occur in maternal vitamin D deficiency (see previous discussion) and also where the mother has untreated hypoparathyroidism. As in primary hyperparathyroidism, the bones are rarefied and spontaneous fractures may occur. Biochemically the condition evolves from hypocalcemia (present prenatally and responsible for the condition) through normocalcemia to hypercalcemia caused by an autonomous release of PTH even when the neonate is receiving an adequate oral calcium intake. Hypophosphatemia may be caused by PTH-induced phosphaturia. During the first few months of life, the skeletal lesions tend to heal rapidly and the parathyroid glands regress and resume normal function.
Clinical Manifestations and Complications
These infants are always hypercalcemic; soon after delivery they become hypophosphatemic. Immunoreactive PTH levels are high. Aminoaciduria, probably caused by a direct effect of PTH on the renal tubule, is common. Erosion of bone may be seen on skeletal radiographs.
Treatment of Neonatal Hypercalcemia
Hypercalcemia can lead to gastrointestinal dysfunction, failure to thrive, hypercalciuria, and, when severe, dehydration. Treatment should be tailored to fit the severity of the hypercalcemia and its consequences. Emergency treatment is needed for dehydration. Rehydration with 0.9% saline corrects the dehydration of hypercalcemia and induces a kaliuresis. Urinary calcium losses can be further increased by the administration of furosemide 6 mg/kg/day (in divided doses). Calcitonin, given subcutaneously, may be effective for a few days, but tolerance to its action soon develops. In theory, bisphosphonates, which have been used to treat hypercalcemia in older children, might prove useful in this type of emergency. Corticosteroids are not beneficial in hypercalcemia due to hyperparathyroidism, but are effective in Williams syndrome and vitamin D toxicity. Clearly, their chronic use should be avoided because of the many adverse consequences of glucocorticoid administration to young children. In neonatal hyperparathyroidism, emergency parathyroidectomy has been used as a definitive therapy with, in some cases, autotransplantation of some of the parathyroid tissue into an accessible site such as the forearm.
Milder degrees of chronic hypercalcemia can be treated with a low-calcium diet. This should not be undertaken without expert nutritional advice because both calcium depletion and phosphate depletion can result from injudicious use of such diets in growing children. Where hypercalcemia is mild, especially in the aptly named familial benign hypercalcemia (familial hypocalciuric hypercalcemia), no therapy is needed.
Neonatal Hypocalcemia
Neonatal hypocalcemia is generally divided into early and late forms according to the age (in days) of onset. The early form generally begins on the first to third day of life, while the late form becomes evident only after a week or so.
Early Neonatal Hypocalcemia
Hypocalcemia occurs in low-birth-weight and sick infants 1 to 4 days of age. Hypocalcemia may present with jittery movements, convulsions, apnea, or myocardial dysfunction. It may be considered an exaggeration of the physiologic drop in ionized calcium seen at this age.
Pathophysiology
The causes of this hypocalcemia, which is more marked in preterm infants, are unknown. Parathyroid underactivity secondary to the normally high serum calcium of the fetus, diminished bone resorption associated with high calcitonin levels, and poor dietary calcium intake and absorption have all been proposed.
Diagnosis
Direct measurement of ionized calcium has been advocated because unlike measures of total serum calcium, no correction needs to be made for protein concentration or blood pH. Electrocardiography (long QT interval) may also provide a useful clue to the presence of subnormal ionized calcium. An ionized calcium concentration less than 2.5 mg/dL can lead to clinical symptoms. Although theoretically attractive, in practical neonatal intensive care unit settings the ability to measure ionized calcium directly does not seem to bring much clinical advantage over measurement of total calcium.
Late Neonatal Hypocalcemia
This disorder presents clinically at 5 to 10 days of age in full-term, and apparently healthy, neonates. Hypocalcemia is associated with elevated serum phosphate levels. The causes and biochemical findings are summarized in Table 64.1.
Permanent Hypoparathyroidism
Primary hypoparathyroidism may be due to inherited or sporadic isolated absence of the parathyroid glands. In some cases, hypoparathyroidism is combined with some or all of the other features of the DiGeorge syndrome (thymic aplasia, severe congenital heart disease with conotruncal malformations, and certain facial abnormalities). The effect of hypoparathyroidism is to reduce the capacity of affected infants to regulate serum calcium and to excrete phosphate. Symptomatic hypocalcemia may occur within the first day or two after delivery but, more commonly, it appears only as serum phosphate levels rise at 6 to 9 days of age.
Epidemiology
The DiGeorge syndrome is one of a group of overlapping syndromes associated with microdeletions of chromosome 22q and is considered to be a contiguous gene syndrome. Some cases are dominantly inherited. The severity and duration of the hypoparathyroidism are quite variable. It may resolve clinically after the neonatal period, sometimes recurring at times of physical stress such as cardiac catheterization or surgery.
TABLE 64.1. CAUSES OF LATE NEONATAL HYPOCALCEMIA AND ASSOCIATED BIOCHEMICAL FINDINGS | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


