Fig. 1
Right and left humeri of a patient with osteogenesis imperfecta
Deformities of the radius and ulna occur less frequently than deformities of the humerus with each forearm bone involved in 18 % and 19 % of patients, respectively (Amako et al. 2004). No matter what upper extremity bone is studied, the magnitude of angular deformity correlates with the severity of the disease phenotype (Amako et al. 2004); thus, the most pronounced upper extremity deformities were seen in the patients with type 3 OI.
In another study of 489 OI patients, radial head subluxation or dislocation was found in 83 patients (17 %) (Fassier et al. 2007). Radial head deformity correlated to severity of disease with the radius dislocating either posteriorly or posterolaterally. Patients with type V OI seem particularly prone to radial head deformity (Fassier et al. 2007).
Deformity of the upper extremity has functional implications. In 159 children given the PEDI outcome tool, mean self-care and mobility scores were negatively correlated to the amount of angular deformity as measured on the radiographs (Amako et al. 2004). Despite this functional loss, upper extremity surgery is performed much less frequently than the correction of deformity in the lower extremities of OI patients (Root 1981).
Diagnostic Evaluation
Osteogenesis imperfecta is most often diagnosed by sequencing for gene mutations in patients who have a suspicious phenotype. An avulsion fracture of the olecranon has been described as being pathognomonic for osteogenesis imperfecta (Stott and Zionts 1993). Occasionally, skin fibroblasts are obtained from biopsy specimens and studied; however, this more traditional means of making a diagnosis is required much less often as the specific genetic makeup of OI is now better understood. Imaging can include a skull film for wormian bones. Most OI patients are imaged primarily with plain films of areas of bone deformity. Child abuse is often a possible diagnosis in children with frequent fractures and distinguishing children with brittle bones from those who are subject to frequent trauma can be a difficult task.
Medical Treatment
Bisphosphonates are analogs of pyrophosphate that bind to the hydroxyapatite crystals in the bone matrix. Pamidronate, the first bisphosphonate used for the treatment of OI, has been shown to decrease the number of fractures and increase bone mass using meta-analysis techniques combining data from multiple randomized clinical trial (Phillipi et al. 2008). Bisphosphonates can have some serious toxicity and clinical trials continue to be conducted to determine when to initiate treatment, what is the optimal dosage and route of administration, and how much total drug should be given. Among reported complications of bisphosphonates is an increase in nonunion at the site of osteotomies, but not an increased rate of fractures (Munns et al. 2004). Therefore, thoughtful planning as to the administration of a bisphosphonate around the time of surgery should be discussed as a team and perhaps the drug held until there is radiographic callus visible at the osteotomy site.
Surgical Treatment
Humeral rodding can be approached by an anterolateral incision over the apex of the main deformity. The radial nerve must be identified and protected and in cases of severe deformity may not be in the expected anatomic location. Multiple osteotomies may be required to straighten the segment. A telescopic or solid rod can be placed from proximal to distal (Fig. 2). The rod should be directed down the medial column of the distal humerus, if possible, to account for the recurrence of varus with growth of the patient (Fig. 3; Root 1981). Postoperative management consists of a sling and swathe that can be removed when the patient is comfortable. As in lower extremity treatment, the need for additional rodding procedures is often necessary as the deformity tends to return over time, particularly in growing children (Amako et al. 2004; Root 1981).
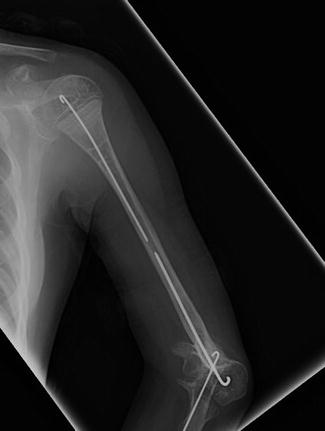
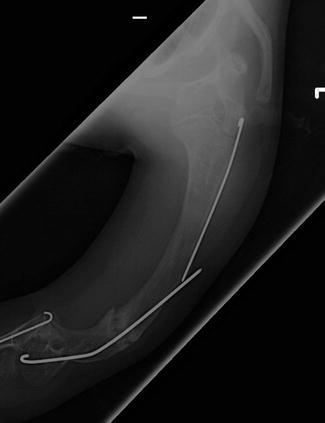
Fig. 2
Postoperative humerus radiograph after intramedullary rodding
Fig. 3
Recurrent varus deformity after humeral rodding
Rodding the forearm can be difficult as the patients who require this procedure are likely to have severe deformity coupled with canals that are so small and deformed that placing a rod inside the canal is inherently challenging. Rod choice is limited to solid rods in the forearm. Root et al. suggest osteotomies of the radius and ulna with rodding of the ulna only (Root 1981). The forearm should be protected in a splint for a few weeks after surgery. In several series, rodding of the bones of the forearm was found to have poor results and caution was advised when considering the procedure. However, most of these articles predate combined medical and surgical management and newer studies are required to determine if surgery is helpful once the biology of the bone is altered.
Rickets
Introduction
Rickets is the result of decreased mineralization of trabecular bone and is characterized by growth retardation and bony deformity. Histologically, an increase in the amount of unmineralized osteoid is the hallmark of the disease. Rickets is a disease exclusively of childhood as it affects the growing physis. Osteomalacia is the adult counterpart and occurs after the cessation of growth. There are a number of causes of rickets all of which affect bone homeostasis. Bone mineralization requires tight regulation of calcium and phosphate. As such, any number of disorders that interrupt the homeostasis of calcium and phosphate may result in rickets. Historically, the most commonly encountered form of rickets was the result of vitamin D deficiency. The most commonly encountered form of inherited rickets is X-linked hypophosphatemic rickets . Other forms of rickets occur as a result of vitamin D resistance, defects in phosphate metabolism, and renal abnormalities.
Vitamin D Deficiency Rickets
Introduction
Vitamin D deficiency rickets has decreased in incidence with the increased use of dietary vitamin D supplementation. However, cases still occur and can lead to considerable morbidity. Infants who are exclusively breastfeed and whom do not receive vitamin D supplementation are at increased risk. In addition, children living at northern latitudes, who have decreased sun exposure either for cultural reasons or because of geography, have darker skin, and those fed a strict vegan diet are also at increased risk. A recent increase in the use of sunblock to prevent skin cancer has had as an unintended consequence a decrease in vitamin D production.
Pathophysiology
Vitamin D is obtained from two sources. Vitamin D3, cholecalciferol, is produced in the skin after sun exposure. Alternatively, vitamin D2, ergocalciferol, is obtained from the diet. Both cholecalciferol and ergocalciferol undergo similar processes to achieve their active state and have equivalent biologic potency. Initially, vitamin D is hydroxylated in the liver to 25-hydroxyvitamin D. 25-hydroxyvitamin D undergoes a second hydroxylation in the kidney to its active form, 1,25-dihydroxyvitamin D. 1,25-dihydroxyvitamin D increases absorption of calcium from the gastrointestinal tract and increases resorption of calcium and phosphorus from bone. The net effect is increased serum calcium. As serum calcium levels fall, parathyroid hormone (PTH) levels increase in an effort to maintain normal serum calcium levels. As a consequence of increased circulating PTH, urinary excretion of phosphate increases and serum concentrations of phosphate decrease. With decreasing levels of calcium and phosphate, PTH promotes calcium loss from the bone. As this severity of vitamin d deficiency increases, rachitic bone changes become readily apparent on radiograph and clinical manifestations soon follow.
Clinical Manifestations
Vitamin D deficiency rickets has a wide spectrum of presentations. The most common piece of patient history is failure to supplement vitamin D during breastfeeding (96 %). A recent review of cases of nutritional rickets over a 10-year period found that 81 % of cases occurred in African American patients, 14 % occurred in patients of Arab descent, and only 2 % occurred in Caucasian patients (Lazol et al. 2008). Most cases presented during fall, winter, or spring, while only 14 % of cases occurred during the summer months.
On evaluation, affected children may demonstrate development delay, decreased growth, and failure to thrive. 47 % and 31 % of patients are below the fifth percentile for height and weight, respectively (Lazol et al. 2008). Genu varum of the legs is not uncommon, occurring in 58 % of patients. Other clinical findings include frontal bossing, rachitic rosary, scoliosis, ligamentous laxity, and hypotonia. Patients may present with seizures and tetany as a result of hypocalcemia (Joiner et al. 2000). Vitamin D also plays a role in one’s ability to mount an immune response. An increased rate of pneumonia in patients with rickets has been observed (Muhe et al. 1997). Other patients may come to clinical attention after incidental radiographic findings lead to a diagnosis of rickets. The most common presenting sign is wide swollen joints (74 %) (Lazol et al. 2008).
Radiographic evaluation will demonstrate typical changes at the distal metaphyses of long bones, most often the distal radius and ulna (Fig. 4). These changes include metaphyseal widening, cupping, and fraying of the metaphysis (Chavhan et al. 2010). Repeat radiographs as soon as 2–3 weeks after the initiation of treatment can demonstrate improvement. Other radiographic features commonly seen include rib flaring, widespread osteopenia, and multiple fractures in various stages of healing. This last feature may lead to a diagnostic dilemma with child abuse and osteogenesis imperfecta also included in the differential.
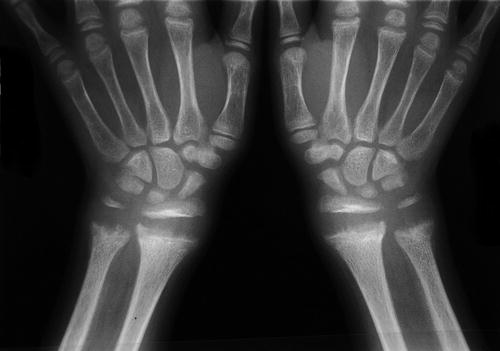
Fig. 4
Typical radiograph of the distal radius and ulna in a patient with vitamin D deficiency rickets (Courtesy of Michael Meagher, MD, Shriners Hospital for Children Honolulu)
The diagnostic evaluation should include serum calcium, phosphorus, PTH, and alkaline phosphatase concentrations. Renal function should be evaluated to rule out other causes of rickets. Finally, 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels should be obtained. The laboratory findings will demonstrate decreased 25-hydroxyvitamin D, normal or increased 1,25-dihydroxyvitamin D, increased PTH, and decreased calcium and phosphorus. The alkaline phosphatase level will be increased in 98 % of cases (Lazol et al. 2008).
Treatment
The first priority of treatment of vitamin D-deficient rickets is to prevent the complications of hypocalcemia, seizures and tetany. Initially, treatment involves replacement of calcium. The second goal of treatment is to replenish vitamin D levels leading to improvement in calcium absorption from the gastrointestinal tract. Daily treatment with vitamin D should be continued until serum alkaline phosphatase levels have returned to normal and radiographic evidence of disease has resolved. During this time, oral calcium supplementation is recommended. In general, the prognosis for patients treated for vitamin D deficiency rickets is good. Bony deformities slowly improve over the course of months to years. In severe cases of genu valgum or varum, severe upper extremity deformity, and short stature, the deformities may not resolve. A review of the literature has not produced evidence for the surgical management of patients with residual upper extremity deformity.
Preventing rickets is an important and achievable goal. Emphasizing the importance of vitamin D supplementation to breastfeeding mothers can decrease the incidence of rickets. The American Academy of Pediatrics in its 2008 recommendations states that children from infancy to adolescence should receive 400 IU of vitamin D daily. In a recent study, only 11–25 % of infants less than 11 months of age were meeting the daily requirement for vitamin D (Perrine et al. 2010). This emphasizes the need for continued improvement in vitamin D supplementation.
Hypophosphatemic Rickets
Introduction
Hypophosphatemic rickets is an inherited disease characterized by rickets, increased renal clearance of phosphate, and short stature. The disease is also known as vitamin X-resistant rickets and is most commonly inherited as an X-linked dominant mode of transmission. The X-linked dominant form of hypophosphatemic rickets accounts for 80 % of cases of hypophosphatemic rickets. Hypophosphatemic rickets can also be inherited as an autosomal dominant disease with reduced penetrance and in a rare autosomal recessive form. The disease occurs more frequently in females (70 %) than males (30 %) (Bhadada et al. 2010). The incidence of hypophosphatemic rickets is 1 per 21,000 people.
Pathophysiology
X-linked hypophosphatemic rickets is caused by a mutation in the phosphate-regulating gene with homologies to endopeptidases on the X chromosome (PHEX). In 87 % of familial cases of hypophosphatemic rickets and 72 % of sporadic cases, a mutation in the PHEX gene is found (Gaucher et al. 2009). The mutation in PHEX leads to its inactivation, which alters phosphate homeostasis. Approximately 80–85 % of phosphate is resorbed in the kidney (Santos et al. 2012). In its normal state, the PHEX gene product decreases the synthesis of fibroblast growth factor 23 (FGF-23). FGF-23 is produced by osteocytes, osteoblasts, and odontoblasts. An increased circulating level of FGF-23 is directly responsible for both phosphaturia and decreased 1-alpha hydroxylation of 25-hydroxyvitamin D (Aono et al. 2009). FGF-23 acts on the proximal renal tubule to downregulate the sodium phosphate cotransporter. Interestingly, cortical bone mineral density seems to be decreased while trabecular bone mineral density seems to be increased in hypophosphatemic rickets (Cheung et al. 2013).
Clinical Features
X-linked hypophosphatemic rickets has a mean age at diagnosis around 1 year of age (Patzer et al. 1999). The most common clinical features of X-linked hypophosphatemic rickets are growth disturbances causing disproportionate short stature, rickets leading to lower extremity deformities, and muscle weakness. Interestingly, the serum phosphate levels are similar in males and females. However, the bony deformities are more severe in males. The growth abnormalities seen in hypophosphatemic rickets are variable and cases of normal adult height have been reported (Santos et al. 2012). Leg length is more severely affected than either trunk length or arm length (Živičnjak et al. 2010). At presentation, the average stature of children with hypophosphatemic rickets is 2.48 standard deviations below controls. Arm length and leg length are 1.81 and 2.90 standard deviations below the normal population, respectively (Zivicnjak et al. 2010). This disturbance in growth appears to worsen during adolescence. The patients may also have abnormalities of their teeth. Many patients present with pathologic fractures and approximately half of patients will have bone pain.
The laboratory evaluation of patients with X-linked hypophosphatemic rickets demonstrates a number of abnormalities. Hypophosphatemia is universally found. Inappropriately increased levels of phosphate are found in the urine despite the hypophosphatemia. The patients will exhibit decreased or inappropriately normal levels of 1,25-dihydroxyvitamin D. FGF-23 levels are elevated. Alkaline phosphatase may be elevated or normal and serum calcium is usually normal.
Treatment
The treatment of X-linked hypophosphatemic rickets is usually with high-dose oral phosphate, calcium, and 1,25-dihydroxyvitamin D supplementation. This approach will allow the rickets and hypophosphatemia to improve. However, it does not fully correct the growth abnormalities and has been associated with nephrocalcinosis and secondary hyperparathyroidism in a considerable number of patients. Approximately 50 % of treated patients develop nephrocalcinosis. This is not seen in untreated patients (Kooh et al. 1994). The hyperparathyroidism in hypophosphatemic rickets can lead to a number of complications including arterial hypertension, ectopic cardiac calcification, and other cardiac abnormalities (Schmitt and Mehls 2004).
Interestingly, treatment of hypophosphatemic rickets with calcitriol and high-dose phosphate leads to an increase in serum FGF-23 levels (Imel et al. 2010). Treatment starting at younger ages has been shown to improve the patient’s growth parameters at skeletal maturity and decrease the radiographic findings associated with rickets (Makitie et al. 2003). In an effort to improve longitudinal growth, treatment with growth hormone supplementation has been attempted. A recent randomized controlled trial demonstrated increased growth without progression of bowing deformities or body disproportion in patients treated with growth hormone (Zivicnjak et al. 2011).
The surgical treatment of hypophosphatemic rickets is aimed at correction of deformities caused by the disease. The majority of these occur in the lower extremities. Early medical treatment has been shown to decrease the need for future surgical intervention (Evans et al. 1980). Of note, the ability to heal osteotomies and fractures may be impaired in patients with hypophosphatemic rickets. As in other metabolic bone diseases, surgical intervention is best preformed as near to skeletal maturity as possible in order to decrease the likelihood of recurrence. The recurrence rate after surgery has been reported to be as high as 90 % (Petje et al. 2008). Both the correction of angular deformity and lengthening procedures have been successfully performed (Fucentese et al. 2008). In one small series of 8 patients undergoing 28 surgeries, there were no cases of nonunion (Fucentese et al. 2008). Fassier-Duval rods have been used in conjunction with Ilizarov frames to correct lower extremity deformity in patients with hypophosphatemic rickets. In a small series of six operations, 50 % of cases had complications (Birke et al. 2011). Although no series exist on the treatment of upper extremity deformity in this condition, experience with care of the lower extremities speaks to the difficulty of treating bony deformity in patients with metabolic bone disease and serves as a reminder to undertake surgical intervention judiciously.
Vitamin D-Dependent Rickets Type I
Introduction
Vitamin D-dependent rickets type I (VDDR-I) is a rare genetic disease caused by a mutation in the 1α-hydroxylase gene. The disease is inherited in an autosomal recessive fashion.
Pathophysiology
Patients with VDDR-I have an inherited defect in vitamin D metabolism. 1,25-dihydroxyvitamin D is the active form of vitamin D and is a key regulator of calcium and phosphate metabolism. The precursor to the active form of vitamin D is made in the skin by the effect of ultraviolet light on 7-dehydocholesterol. Subsequently, vitamin D undergoes two successive hydroxylations to form 1,25-dihyroxyvitamin D. The first occurs in the liver and the second occurs in the proximal renal tubule of the kidney. The genetic defect in VDDR-I is most often a loss of function mutation in the 1α-hydroxylase gene. 1α-hydroxylation is the rate-limiting step in the formation of 1,25-dihydroxyvitamin D. Subsequently, patients are unable to produce the active metabolite of vitamin D. The 1α-hydroxylase enzyme is a member of the cytochrome P450 family.
Clinical Features
Patients with VDDR-I typically present with failure to thrive, muscle weakness, and symptoms of hypocalcemia. They may present with seizures. They often have skeletal changes typical of rickets including genu varum or valgum, rachitic rosary, and pathologic fractures. Finally, they may have secondary hyperparathyroidism. As this is a genetic disorder, the patients typically present in infancy.
Laboratory findings will allow the diagnosis to be elucidated. The patients will have low or no detectable serum 1,25-dihydroxyvitamin D with normal to increased levels of 25-hydroxyvitamin D levels. Calcium and phosphate levels will be decreased. Conversely, parathyroid hormone and alkaline phosphatase levels will be elevated.
Treatment
VDDR-I is treated with the replacement of 1,25-dihydroxyvitamin D in physiologic doses. This has been shown to cause remission of the disease including correction of the laboratory parameters and reversal of the rachitic bony changes.
Vitamin D-Dependent Rickets Type II
Introduction
Vitamin D-dependent rickets type II (VDDR-II) is a rare autosomal recessive disorder caused by a mutation in the vitamin D receptor. The disease results in end-organ resistance to 1,25-dihydroxyvitamin D. VDDR-II is further classified by the presence (type IIA) or absence of alopecia (type IIB).
Pathophysiology
VDDR-II is caused by a mutation in the vitamin D receptor gene found on chromosome 12 (Malloy et al. 1990; Feldman et al. 1982). The disease is inherited in an autosomal recessive fashion. Several mutations have been described leading to variability in the patients’ response to 1,25-dihydroxyvitamin D. The most common mutations result in a defective steroid binding domain in the vitamin D receptor (Malloy et al. 1990). These patients have no detectable 1,25-dihydroxyvitamin D activity.
Clinical Features
The onset of disease is usually early in infancy. The patients have a severe rickets diathesis and present with severe bony changes and symptoms of hypocalcemia. A distinguishing feature of VDDR-II is the associated finding of total scalp and body alopecia. This is seen in a significant proportion of patients but not universally. Patients may be born with hair and later go on to develop alopecia (Al-Khenaizan and Vitale 2003). Importantly, patients with alopecia may have worse disease phenotype than those without alopecia. The patients may also exhibit signs and symptoms of immunodeficiency (Soni et al. 2008). Although the exact mechanism linking active vitamin D and the hair follicle is unknown, the vitamin D receptor is found in the dermal papillae of the hair follicle as well as in keratinocytes of the outer root sheath.
The diagnosis can be made based on the clinical history and laboratory evaluation. The patients have similar laboratory abnormalities as VDDR-I with one exception. The sine qua non is an elevated level of 1,25-dihydroxyvitamin D.
Treatment
The treatment of VDDR-II involves replacement with either high-dose vitamin D analogs or calcium supplementation (Kruse and Feldmann 1995). VDDR-II varies in its responsiveness to vitamin D supplementation. In some cases, intravenous calcium replacement may be required to reverse clinical symptoms. Interestingly, treatment of VDDR-II does not reverse the associated alopecia.
Osteopetrosis
Introduction
Osteopetrosis is an uncommon genetically inherited disease resulting in abnormally dense bone. The entity is also known as marble bone disease or Albers-Schonberg disease. Its incidence ranges from 1 in 250,000 live births in the autosomal recessive form to 5 per 100,000 live births in the autosomal dominant form. The autosomal recessive forms tend to result in more severe disease and, if untreated, are fatal in infancy.
Pathophysiology
Osteopetrosis varies substantially in severity between different types of the disease. This variation is a reflection of the multiple different genetic etiologies. At least 10 different genes have been implicated in the pathogenesis of osteopetrosis. Mutations in T-cell immune regulator 1, RANK, RANKL, CLCN7 (a chloride channel), and carbonic anhydrase II, among others, have been described (Michou and Brown 2011). All of these genes in one way or another impact the function of osteoclasts, ultimately leading to dysregulation of bone homeostasis. Osteoclasts are multinucleated cells that originate from mononuclear precursors. Loss of function mutations in RANK and RANKL leads to the rare osteoclast-deficient form of osteopetrosis (Guerrini et al. 2008). The more common osteoclast-rich forms of osteopetrosis are most frequently a result of mutations in CLCN7. Loss of this chloride channel function prevents acidification of the resorption lacuna. Subsequently, bone demineralization is significantly impaired. Interestingly, only 70 % of cases of osteopetrosis are explained by a currently known mutation (Stark and Savarirayan 2009).
Clinical Features
Osteopetrosis is characterized by increased bone density leading to a variety of clinical problems. The disease can range from fatal in infancy to asymptomatic. The more severely affected patients have macrocephaly and other craniofacial abnormalities. Of particular importance are the systemic changes that occur as a result of the disease. The bone marrow is replaced by bone leading to anemia and immunodeficiency. A commonly seen feature of many sclerosing bone diseases is cranial nerve compression secondary to stenosis at neuroforamina. This is also seen in osteopetrosis. The skeletal findings are common among all subtypes of osteopetrosis though their severity varies. The skeleton shows increased bone density with diffuse sclerosis; there can be modeling deformities at the metaphysis, a predisposition to pathologic fractures, and osteomyelitis. The most common modeling defect is the “Erlenmeyer flask” seen at the distal femur. Another common radiographic feature is the bone in bone appearance of the vertebrae, ilium, and phalanges. Patients are at risk for dental caries.
There are three main types of osteopetrosis: autosomal recessive osteopetrosis, intermediate osteopetrosis, and autosomal dominant osteopetrosis. The subtypes will be discussed in the following paragraphs.
Autosomal recessive osteopetrosis, sometimes known as malignant osteopetrosis, becomes clinically apparent in the first few weeks to months of life. It is a life-threatening disease. Pancytopenia leads to profound immunodeficiency and extramedullary hematopoiesis occurs in the liver and spleen. Subsequently, hepatosplenomegaly is present in most patients (Del Fattore et al. 2006). The patients have an increase in bone quantity, but the bone is brittle, leading to pathologic fractures. Patients may also have macrocephaly and frontal bossing. Cranial nerves are entrapped at their foramina and 78 % of patients have hearing loss. In addition, patients may present with hydrocephalus secondary to outflow obstruction in the posterior fossa (Al-Tamimi et al. 2008). Laboratory evaluation is positive for hypocalcemia. The patients may have symptoms related to their hypocalcemia including seizures and hyperparathyroidism. Patients with the autosomal recessive form of the disease who are untreated have a shortened lifespan.
There are several subtypes of autosomal recessive osteopetrosis. The first is neuropathic, which is characterized by neurodegeneration and seizures despite normal calcium levels. A second subtype is associated with renal tubular acidosis and has a less severe clinical course (Cotter et al. 2005). In this rare form of the disease, profound hematopoietic deficiencies are uncommon.
An intermediate form of osteopetrosis has been described. Similar to infantile osteopetrosis, this disease is inherited in an autosomal recessive manner. However, the disease has its onset in childhood. The children may have some disturbance in hematopoiesis, but it is usually mild. The other features of the disease tend to be much less severe than that seen in infantile osteopetrosis.
The last form of osteopetrosis is the autosomal dominant form, which presents in adolescence. The disease has variable penetrance with 90 % of all patients with known mutations demonstrating the phenotype. In autosomal dominant osteopetrosis, most findings are related to the skeleton. The patients may develop hip osteoarthritis, pathologic fractures, scoliosis, and dental caries. Only 5% of affected people have hearing or visual loss. Unlike the autosomal recessive forms of osteopetrosis, patients typically have a normal life expectancy. The diagnosis of autosomal dominant osteopetrosis can be confirmed with an elevated tartrate-resistant acid phosphatase serum level (Waguespack et al. 2002).
Treatment
The treatment of osteopetrosis is largely aimed at treating the complications of the disease. No completely effective medical treatment has been developed to date. High-dose steroids have been used with mixed results. Interferon gamma has been used with some success in patients with the infantile malignant form of the disease (Key et al. 1995). Interferon gamma has been shown to decrease bone density, increase the medullary space, increase the size of neuroforamina, and decrease the rates of infection in patients with osteopetrosis (Driessen et al. 2003). Seizures are managed with calcium and vitamin D supplementation. In cases of symptomatic anemia, the patients may require blood products. Patients should be followed at regular intervals for evaluation of hearing and visional loss. In patients with the malignant autosomal recessive form of osteopetrosis, hematopoietic stem cell transplantation has been used with some success. In the ideal situation with the availability of an identically matched donor, 73 % of patients were disease-free at 5 years (Driessen et al. 2003). The overall survival in one study, including 122 patients who had undergone stem cell transplantation, was 46 % at 5-year follow-up (Driessen et al. 2003). The expected outcome of transplantation is prevention of disease progression not reversal of already apparent disease manifestations. For instance, the rate of vision impairment was 52 % in patients treated at less than 3 months of age, while it was 86 % in patients older than 6 months (Driessen et al. 2003). Few patients demonstrate improvement in vision after stem cell transplantation. As such, stem cell transplantation ideally occurs prior to 3 months of age. The surgical management of complications from osteopetrosis is particularly challenging. Frequently, patients with osteopetrosis have osteoarthritis and pathologic fractures. In cases were fracture fixation is required, nonunion or delayed union is common. A recent report of operative fixation of a humerus fracture treated for nonunion after initial conservative therapy suggests a role for BMP in cases of atrophic nonunions (Rafiq et al. 2009). Intraoperatively, one should expect difficulty with drilling and reaming (Nakayama et al. 2007). Lastly, treating surgeons should be aware that patients with osteopetrosis are at increased risk for infection after surgical procedures.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


