Meconium Disease
Intestinal obstruction is one of the most common admitting diagnoses to the neonatal intensive care unit, accounting for as many as one-third of all admissions.1 Failure to pass meconium within the first 24 to 48 hours of life, feeding intolerance, abdominal distension, and bilious emesis are hallmarks of intestinal obstruction in the newborn, and evoke a differential diagnosis of obstruction based on anatomic, metabolic, and functional considerations. The term meconium disease refers to meconium ileus and meconium plug syndrome. These conditions are considered separately from functional or anatomic causes of neonatal intestinal obstruction, such as Hirschsprung disease, intestinal atresia, and anorectal malformations.
Meconium Ileus
Meconium ileus (MI) is one of the most common causes of intestinal obstruction in the newborn, accounting for 9–33% of neonatal intestinal obstructions.2 It is characterized by extremely viscid, protein-rich, inspissated meconium causing an intraluminal obstruction in the distal ileum, usually at the ileocecal valve. It is often the earliest clinical manifestation of cystic fibrosis (CF), occurring in approximately 16% of patients with CF.3 Although MI can occur with other uncommon conditions such as pancreatic aplasia and total colonic aganglionosis, it is often considered pathognomonic for CF.4,5 MI may be an early indication of a more severe phenotype of cystic fibrosis, as suggested by significantly diminished pulmonary function found in children with a history of MI compared to age- and gender-matched children with CF who did not have MI.6
Due to abnormalities of exocrine mucus secretion and pancreatic enzyme deficiency, the meconium in MI differs from normal meconium. Meconium in MI has less water content (65% vs 75%) when compared to normal meconium, lower sucrase and lactase levels, increased albumin, and decreased pancreatic enzymes.7–9 Additionally, concentrations of sodium, potassium, magnesium, heavy metals, and carbohydrates in MI meconium are reduced in CF. Concentrations of protein nitrogen are increased and composed of abnormal mucoproteins.10–12 Therefore, more viscous intestinal mucus in the absence of degrading enzymes results in thick, dehydrated meconium that obstructs the intestine.13
Cystic Fibrosis
An understanding of CF is important for all clinicians involved in the management of MI patients. CF is the most common, potentially lethal genetic defect affecting Caucasians. Each year 1,200 infants are born with CF (1 : 2500 live births), and 30,000 children and young adults live with CF in the USA.4 It is an inherited autosomal recessive disease with a 4–5% carrier rate.14 The incidence of CF is much lower in non-Caucasian populations: 1 in 10,500 Native American Aleut (Eskimo) births, 1 in 13,500 in Hispanic–Caucasian births, 1 in 15,000 African–American births (much lower in native Africans), and 1 in 31,000 in Asian–American births.
Genetics
In 1989, the CF locus was localized through linkage analysis to human chromosome 7q31, and it was discovered that mutations in the CF transmembrane (conductance) regulator (CFTR) gene result in CF.15–18 The cell membrane protein coded by CFTR is a 3′–5′-cyclic adenosine monophosphate (cAMP)-induced chloride channel, which also regulates the flow of other ions across the apical surface of epithelial cells. The alteration in CFTR results in an abnormal electrolyte content in the environment external to the apical surface of epithelial membranes. This leads to desiccation and reduced clearance of secretions from tubular structures lined by affected epithelia.
The most common mutation of the CFTR gene, F508del (previously known as ΔF508), is a three base-pair deletion that results in the removal of a phenylalanine residue at amino acid position 508 of the CFTR. Although there are currently 1,903 mutations listed in the CFTR database, the F508del mutation is responsible for approximately 70% of abnormal CF genes.15,16,18,19 In families with MI, there is a significantly higher occurrence rate than the expected 25% for an autosomal recessive genetic disorder.20,21 In one series, 79% of CF patients with the F508del mutation presented with abdominal complaints (including MI) rather than pulmonary complaints.4 However, there is no evidence of distinct allelic frequencies or haplotypic variants in CF patients with MI compared with those without22 or in CF patients with significant liver disease.23,24
Gastrointestinal Pathophysiology
Cystic fibrosis is characterized by mucoviscidosis of exocrine secretions throughout the body resulting from abnormal transport of chloride ions across apical membranes of epithelial cells.4,25–27 Abnormal bicarbonate transport also affects mucin formation in CF.28 The clinical result is chronic obstruction and infection of the respiratory tract, insufficiency of the exocrine pancreas, and elevated sweat chloride levels.29 Other clinical variants, such as patients with chronic sinusitis or adult males with congenital bilateral absence of the vas deferens (CBAVD), who typically have little other clinical involvement, have been described (Fig. 32-1).30–32 In patients with CBAVD, the CFTR genotype usually includes at least one mild mutation not typical of CF patients. The mild-mutation allele is frequently associated with a severe mutation on the other allele, such as the F508del mutation.6,33 CBAVD has been described in a patient with F508del and G551D mutations, both of which were categorized as severe.34 The allele G551D is the third most common CF-associated mutation, and patients affected by this mutation may have pancreatic insufficiency, pulmonary symptoms, and an episode of MI equivalent, indicating CBAVD may be associated with a more severe CF phenotype.7,35
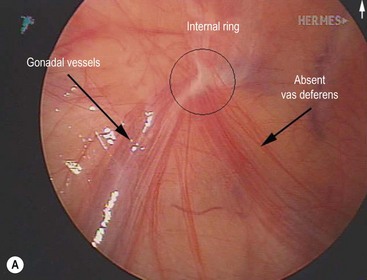
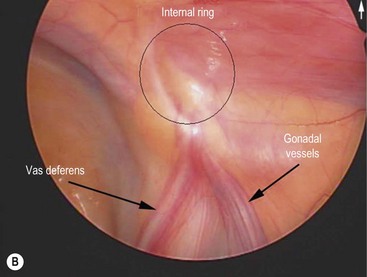
FIGURE 32-1 Congenital bilateral absence of the vas deferens (CBAVD). (A) Laparoscopic view of a patient’s left internal ring. (B) Compare with a laparoscopic view of a similar-aged patient’s right internal ring with a normal vas deferens. (From Escobar MA, Lau ST, Glick PL. Congenital bilateral absence of the vas deferens. J Pediatr Surg 2008;43:1222–3.)
Development of both the pancreas and intestinal tract in fetuses with CF is abnormal. In patients with CF, abnormal pancreatic secretions obstruct the ductal system leading to autodigestion of the acinar cells, fatty replacement of pancreatic parenchyma, and fibrosis. Although this process begins in utero, it occurs variably over time. Regardless, pancreatic insufficiency is prevalent in young infants with CF and has a significant impact on growth and nutrition.23
Pancreatic insufficiency plays a central role in the pathogenesis of MI. Congenital stenosis of the pancreatic ducts is associated with meconium-induced bowel obstruction.36 This is further supported by the fact that two-thirds of infants found to have CF by neonatal screening are pancreatic insufficient at birth.37 However, approximately 10% of patients with CF are pancreatic sufficient and tend to have a milder course. Also, pancreatic lesions are variable at birth and become more severe in CF children older than age 1.38 This finding suggests that pancreatic insufficiency is not the leading cause of abnormal meconium in MI. It appears that a prevalence of intestinal glandular abnormalities contribute more significantly to the production of abnormal meconium.39 The lack of concordance between MI and the severity of pancreatic disease and the preponderance of intestinal glandular lesions implies that intraluminal intestinal factors contribute more to the development of MI than the absence of pancreatic secretions.9,11,12,36–43
Abnormal intestinal motility may also contribute to the development of MI. Some patients with CF have prolonged small intestinal transit times.44,45 Also, the CFTR ion channel defect results in an exocrine secretion that is rich in sodium and chloride which can lead to further dehydration of the intraluminal contents, resulting in impaired clearance.6 Non-CF diseases associated with abnormal gut motility, such as Hirschsprung disease and chronic intestinal pseudo-obstruction, have been associated with MI-like disease, signifying that decreased peristalsis may allow for increased reabsorption of water, thus favoring the development of abnormal meconium.46–48
Prenatal Diagnosis and Screening
The American College of Obstetrics and Gynecology recommends all women of reproductive age should be offered CF carrier screening.14 Based on the results of CF screening, the antenatal diagnosis of MI can be made in two different groups: a high-risk group and a low-risk group. In the low-risk group, the diagnosis is suspected when the sonographic appearances of MI are found on routine prenatal ultrasound in a mother with a negative CF carrier screen. Sonographic findings consistent with MI in a fetus with parents who are known carriers of CF, and pregnancies subsequent to the birth of a CF-affected child, are considered high risk. Parents of a child with CF are considered to be obligate carriers of a CF mutation.
An algorithm has been established which may be useful in decision making and management of the fetus suspected of having MI (Fig. 32-2).49–51 If both parents are carriers, evaluation of the fetus should be made by chorionic villus sampling or amniocentesis. In a pregnancy where CF is suspected, sonographic examinations are performed monthly until delivery. This evaluation allows the early detection of potential complications and prepares the clinicians for special or urgent medical or surgical needs upon delivery.
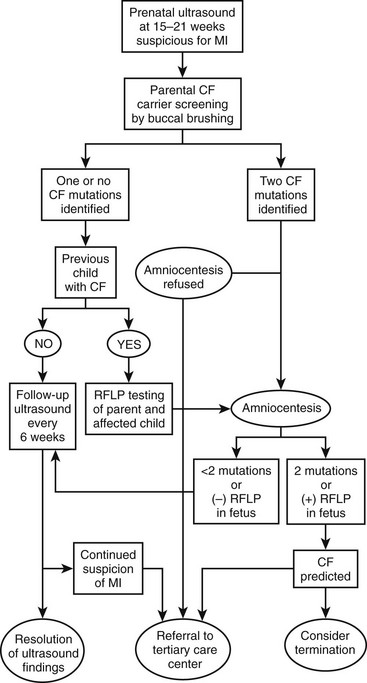
FIGURE 32-2 Suggested algorithm for antenatal management of suspected meconium ileus (MI) and cystic fibrosis (CF). US, ultrasonography; RFLP, restriction fragment length polymorphism. (Adapted from Irish MS, Ragi JM, Karamanoukian H, et al. Prenatal diagnosis of the fetus with cystic fibrosis and meconium ileus. Pediatr Surg Int 1997;12:434–6.)
Sonographic Evaluation
Sonographic characteristics associated with MI include a hyperechoic, intra-abdominal mass (inspissated meconium) (Fig. 32-3), dilated bowel, and nonvisualization of the gallbladder.52 Normal fetal meconium, when visualized in the second and third trimesters, is usually hypoechoic or isoechoic to adjacent abdominal structures.52–57 The sensitivity of intra-abdominal echogenic masses in the detection of MI/CF is reported to be between 30–70%.57 In addition to MI, hyperechoic bowel has been reported with Down syndrome, intrauterine growth retardation, prematurity, in utero cytomegalovirus infection, intestinal atresia, abruptio placenta, and fetal demise.52–55,58–64 The importance of hyperechoic fetal bowel is related to gestational age at detection, ascites, calcification, volume of amniotic fluid, and the presence of other fetal anomalies.57 The positive predictive value of hyperechoic masses in a high-risk fetus is estimated to be 52%, but is only 6.4% in the low-risk fetus.52 It is important to note that hyperechoic bowel has been found to be a normal variant in both the second and third trimesters.56,57,65
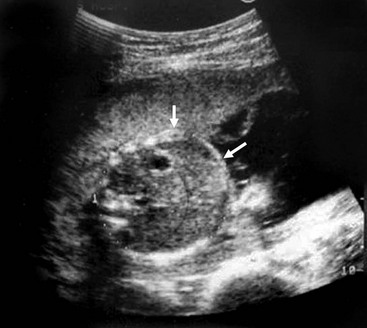
FIGURE 32-3 Ultrasound image of a 22-week gestation demonstrating a 2 cm × 3 cm intraluminal (distal ileum) mass (arrows) consistent with meconium inspissation (meconium ileus). (From Irish MS, Ragi JM, Karamanoukian H, et al. Prenatal diagnosis of the fetus with cystic fibrosis and meconium ileus. Pediatr Surg Int 1997;12:434–6.)
The finding of dilated bowel on prenatal ultrasound (US), in association with a family history of CF, has been reported less frequently than that of hyperechoic bowel. In MI, bowel dilation is caused by obstruction from meconium, but mimics findings in midgut volvulus, congenital bands, intestinal atresia, intestinal duplication, internal hernia, meconium plug syndrome, and Hirschsprung disease.66 The correlation of dilated fetal bowel and MI suggests that dilated fetal bowel warrants parental testing for CF and continued sonographic surveillance of the fetus.
The inability to visualize the gallbladder on fetal ultrasound has also been associated with CF.67 Combined with other sonographic features, nonvisualization of the gallbladder can be useful in the prenatal detection of the disease. However, caution should be exercised in the interpretation of an absent gallbladder as the differential diagnosis also includes biliary atresia, omphalocele, diaphragmatic hernia, chromosomal abnormalities, and a normal pregnancy.
Clinical Presentation
MI is categorized as either simple or complicated. The thickened meconium begins to form in utero. As it obstructs the mid-ileum, proximal bowel dilatation and thickening, along with congestion, occur. Approximately one-half of these neonates present with simple uncomplicated obstruction.4 The remaining patients present with complications of MI, including volvulus, gangrene, atresia, and/or perforation, which may result in meconium peritonitis and giant cystic meconium peritonitis.68–73
Simple Meconium Ileus
In simple MI, the terminal ileum is filled with firm concretions. The bowel in this area is small in diameter and molds around the inspissated lumps of meconium. The ileum becomes dilated and is filled with thick sticky meconium with gas and fluid found within the small bowel proximal to this area.4 Newborns with uncomplicated MI often appear healthy immediately after birth. However, within 1 to 2 days, they develop abdominal distension and bilious emesis. Normal meconium will not be passed. Eventually, dilated loops of bowel become visible on exam and have a ‘doughy’ character that indent on palpation. The rectum and anus are often narrow, a finding that may be misinterpreted as anal stenosis. The presentation of the baby with MI is similar to many types of neonatal small bowel obstruction. Therefore, the clinician should simultaneously consider malrotation, small intestinal atresia, colonic atresia, and meconium plug syndrome. The history, physical examination, and contrast enema help distinguish between these entities.
Complicated Meconium Ileus
Infants with complicated MI present with symptoms within 24 hours of birth. Some newborns are symptomatic immediately after birth as a result of in utero perforation or bowel compromise. Signs of peritonitis, including distension, tenderness, abdominal wall edema and erythema, and clinical evidence of sepsis may be found on the initial neonatal exam. Abdominal distension can be so severe as to cause immediate respiratory distress. A palpable mass suggests pseudocyst formation, which results from in utero bowel perforation.74,75 The neonate may present in extremis and need urgent resuscitation and operative exploration.
Historically, segmental volvulus was reported to be the most common complication of MI.68,69 Prenatal volvulus of the meconium-distended segment of ileum may lead to interruption of the mesenteric blood flow, which can result in ischemic necrosis, intestinal atresia with an associated mesenteric defect, or perforation. When an in utero perforation occurs, most of the sterile meconium is reabsorbed with trace amounts becoming calcified. Atretic segments are common in MI and the affected bowel may appear viable, showing no evidence of perforation or gangrene. 12–17% of neonates born with jejunoileal atresia have CF.4,76,77 Therefore, all neonates with jejunoileal atresia and an abnormal meconium presentation (MI, meconium plug syndrome, giant cystic meconium peritonitis, etc.) should undergo a sweat chloride test.4
The incidence of CF in neonates with meconium peritonitis is reported to be 15–40%.78 Four types of meconium peritonitis have been recognized including: adhesive meconium peritonitis, giant cystic meconium peritonitis or pseudocyst, meconium ascites, and infected meconium peritonitis.79 In addition to MI, other causes of in utero bowel perforation must also be considered (atresia, stenosis, colonic disorders, imperforate anus) in this clinical setting. The differences in clinical presentation are secondary to the timing of the perforation and whether or not the perforation sealed spontaneously. The site of perforation is usually closed by birth. Not surprisingly, mortality is increased in cases where the perforation remains open.79 Initially, meconium peritonitis is a nonbacterial, chemical, and foreign body peritonitis occurring during gestation. As meconium escapes the obstructed bowel, a sterile chemical peritonitis ensues. After delivery, bacterial superinfection may occur with colonization of the gastrointestinal tract. It is important to note that meconium peritonitis can also occur without MI and is not pathognomonic for CF.4,75,79
Radiographic Features
Simple MI is characterized by a pattern of unevenly dilated loops of bowel on an abdominal radiograph with the variable presence of air–fluid levels.69,80,81 The absence of air–fluid levels is due to the viscosity of the meconium not allowing an air interface with the fluid. As swallowed air mixes with the tenacious meconium, bubbles of gas may be seen. This soap bubble appearance (Fig. 32-4) depends on the viscosity of the meconium and is not a constant feature.71,80,81 While each of these features alone is not diagnostic of MI, collectively with a family history of CF, they strongly suggest the diagnosis.
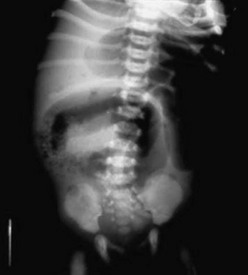
FIGURE 32-4 This abdominal radiograph in a neonate with meconium ileus shows the typical ground-glass appearance in the right lower abdomen. Also note the different-sized loops of distended small bowel.
Radiographic findings in complicated MI vary with the complication. Prenatal ultrasound findings include ascites, intra-abdominal cystic masses, dilated bowel, and calcification.82 Neonatal radiographic findings may include peritoneal calcifications, free air, and/or air-fluid levels (related to atresia).4 Air–fluid levels may be minimally present or absent, misleading the clinician to make an incorrect diagnosis of uncomplicated MI. Speckled calcification on abdominal plain films is highly suggestive of intrauterine intestinal perforation and meconium peritonitis. Radiographic findings of obstruction and a large dense mass with a rim of calcification imply a pseudocyst (Fig. 32-5). These calcium deposits are linear and course along the parietal peritoneum and serosal surface of the visceral organs.83 Interestingly, one-third of cases of complicated MI have no radiologic findings that suggest a complication.84
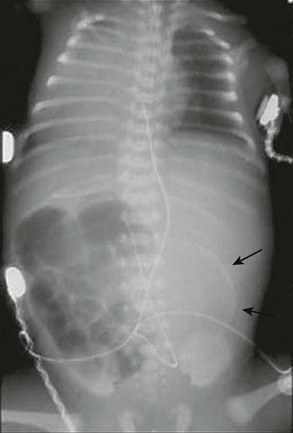
FIGURE 32-5 This neonate presented with evidence of meconium peritonitis. A mass effect in the left abdomen with a rim of calcification (arrows) implies in utero perforation and a pseudocyst.
A contrast enema should be performed in all cases of low intestinal obstruction in the newborn. We advocate an initial water-soluble contrast enema for both diagnosis and treatment. In MI, contrast instillation is monitored fluoroscopically and demonstrates a colon of small caliber, described as the ‘microcolon of disuse,’ often containing small, inspissated rabbit pellets (scybala) of meconium (Fig. 32-6). The enema also identifies cecal position, indicating whether malrotation is present. In complicated cases, such as atresia, a microcolon with reflux into a decompressed terminal ileum may be noted.4 If contrast cannot be refluxed into the dilated small bowel, operative exploration is required for diagnosis and therapy.
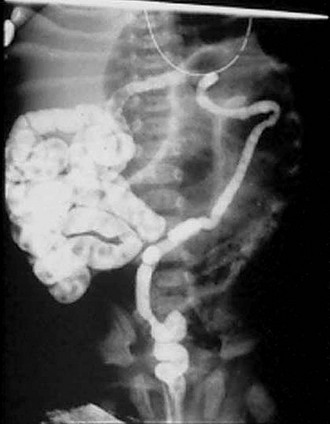
FIGURE 32-6 Classic radiographic findings of meconium ileus are seen on this retrograde contrast study. First, a ‘microcolon of disuse’ is seen. The colon is extremely small and unused. Second, inspissated pellets (filling defects) of meconium are seen in the more proximal small bowel. Third, note there is a small bowel obstruction as the contrast material has not reached the markedly dilated loops of small bowel.
Diagnostic Testing
The diagnosis of CF is established with a sweat test. A sodium concentration of 60 mmol/L in 100 mg of sweat is diagnostic of CF with 40–60 mmol/L being intermediate (but more likely to be diagnostic in infants) and less than 40 mmol/L being normal.85 The test is typically performed at several weeks of life to obtain an adequate sample size. Neonatal CF screening programs using the Guthrie blood spot test for raised concentrations of immunoreactive trypsinogen is available in many countries, but must be confirmed in a two-stage approach incorporating CFTR mutation analysis.86,87 Genetic testing for CFTR mutations is available, however, commercial assays test for a limited number of mutations. Most regional laboratories will provide the results for the four or five most common mutations for the relevant ethnic group or geographical region in their area using the amplification refractory mutation system (ARMS) technique. Stool analyses for albumin, trypsin, and chymotrypsin are available, and abnormal values coupled with operative findings suggest CF.37
Neonates with MI who fail to respond to nonoperative measures may be treated by appendectomy and irrigation with water-soluble contrast into the small bowel via the small bowel or appendiceal stump.88 The appendix (or other intestinal biopsy) may be sent for histologic analysis. Pathognomonic findings or histology for CF include goblet cell hyperplasia and accumulated secretions within crypts or lumen.89
Nonoperative Management of Simple Meconium Ileus
Neonates should initially be managed as any other newborn with intestinal obstruction. This management should include volume resuscitation and ventilator support as necessary. Gastric decompression to prevent progressive abdominal distension, aspiration, and pulmonary compromise is important. In addition, correction of any coagulation disorders and empiric broad-spectrum antibiotic coverage should be initiated.
Under fluoroscopic control, the water-soluble contrast material is slowly infused at low hydrostatic pressure through a catheter inserted into the rectum. Inflation of the catheter balloon should be avoided to minimize the risk of perforation. Upon completion, the catheter is withdrawn and an abdominal radiograph is obtained to evaluate for perforation. The infant is then returned to the neonatal care unit for intensive monitoring and fluid resuscitation. Usually there is rapid passage of meconium pellets followed by semi-liquid meconium, which continues in the ensuing 24–48 hours. Upon instillation of the enema, extraluminal fluid is drawn into the intestinal lumen, hydrating and softening the meconium mass. Warm saline enemas containing 1% N-acetylcysteine (Mucomyst; Apothecon, Princeton, New Jersey) may be given to help complete the evacuation.73 Radiographs should be obtained as clinically indicated to confirm evacuation of the obstruction and to exclude late perforation. If evacuation is incomplete, or if the first attempt at contrast enema evacuation does not reflux contrast into dilated bowel, a second enema may be necessary. However, if progressive distension, signs of peritonitis, or clinical deterioration occur, operative exploration is indicated. After two failed attempts at nonoperative water-soluble enemas, operative intervention is likely warranted.
Following successful evacuation and resuscitation, 5 mL of a 10% N-acetylcysteine solution may be administered every six hours through a nasogastric tube to liquefy the upper gastrointestinal secretions. Feedings with supplemental pancreatic enzymes for those infants confirmed with CF may be initiated when signs of obstruction have subsided. In the past, the success rate of patients with uncomplicated MI, treated with Gastrografin® enemas, has ranged between 63–83%.90,91 However, more recent reports indicate a much lower success rate likely secondary to the use of isotonic enema fluid.4,92
Several potential complications exist with the use of enemas in treating MI. The risk of rectal perforation can be avoided by careful placement of the catheter under fluoroscopic guidance and by not inflating the balloon-tipped catheter. A 23% perforation rate has been demonstrated in patients when inflated balloon catheters were used, and the risk of perforation increases with repeated enemas.93,94 Late perforation, occurring between 12 and 48 hours following the enema, can occur as well. Potential causes for late perforation include severe bowel distension by fluid osmotically drawn into the intestine or by injury to the bowel mucosa by the contrast medium.94 Lower perforation rates have been reported more recently, possibly related to less aggressive enema attempts and isotonic enema agents.4,92 Hypovolemic shock is a risk when delivering hypertonic enemas. Ischemia caused by overdistension is worsened by hypoperfusion caused by hypovolemia due to inadequate fluid resuscitation.95
Operative Management
Simple Meconium Ileus
At laparotomy, manual evacuation of the inspissated meconium can be aided by intraoperative instillation of 2% or 4% N-acetylcysteine or saline solutions. These fluids can be passed antegrade through a nasogastric tube, retrograde through the appendiceal stump, or directly into the meconium through an enterotomy. A purse-string suture is placed in the antimesenteric wall of the bowel and a red rubber catheter is inserted through a small incision within the purse-string. This is followed by gentle instillation of the solution into the proximal bowel and terminal ileum to avoid perforation. Often the thick tenacious meconium can be removed directly through the enterotomy (Fig. 32-7). The dissolved meconium and pellets can be either removed directly or milked into the colon. It is important that the surgeon avoids exposure of the meconium to the peritoneum. Once the meconium is cleared, the enterotomy or appendiceal stump is closed. If necessary, an indwelling intestinal catheter or a T-tube may be left through the enterotomy for the purpose of postoperative bowel irrigation, decompression, pancreatic enzyme instillation, and/or feeding.96 The enterostomy tube should be positioned at the junction of the proximal dilated bowel and collapsed distal ileum. The irrigations are begun in the early postoperative period and after successful clearance of meconium, the tubes are removed and the enterocutaneous fistula is allowed to close spontaneously.96–100
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


