Mechanisms of Adverse Drug Reactions in Children
Michael Rieder
Bruce Carleton
Adverse Drug Reactions
Children have been among the major beneficiaries of the era of Specific Therapy and the Therapeutic Revolution that started with Dömagk’s description of the antimicrobial activity of Prontosil™ (1). However, as with most major advances, this has come at a price, most notably the fact that potent drugs can have potent adverse events. This was recognized very quickly, in that a number of serious adverse events associated with therapy were described in 1937, the year that sulfonamides first entered the market as antimicrobials (2). In fact, the history of drug regulation has been notable by being driven by therapeutic disasters involving children, the first of which was the Elixir of Sulfanilamide tragedy (2). Since then, it has been increasingly appreciated that adverse drug reactions (ADRs) are common and important complications of therapy that are seen with variable frequency in relation to therapy with essentially all drugs used for the care of children.
The original definition of ADRs by the World Health Organization was “any noxious, unintended, and undesired effect of a drug, which occurs at doses used in humans for prophylaxis, diagnosis, or therapy” (3). Although this is a useful definition, it is somewhat restricted in that it does not include errors in drug administration or dosing, issues that are of special relevance to children. A more useful definition is that an ADR is “an injury resulting from medical intervention related to a drug, which includes errors in administration” (4). This definition, which includes problems related to errors in drug administration, is much more useful with respect to considering ADRs that occur in children.
Adverse Drug Reactions—The Burden
ADRs are one of the most common causes of mortality and serious morbidity in developed countries (2,5,6). ADRs account for 5% of hospital admissions and complicate, at a minimal rate, 5% of the courses of most medication, with the rate climbing much higher among populations such as patients with cancer or in environments such as the critical care unit (5,6). Based on a large study of adverse events associated with therapy, it has been suggested that ADRs are the fourth most common cause of death in Canada and the United States, given that nearly 100,000 Americans a year die as a consequence of ADRs (6). The risk for children is less well defined than that for adults but does appear to be similar overall. Indeed, there may be circumstances in which the risk may be higher for children than for adults (6,7,8,9).
There are well-known risk factors for ADRs (Table 59.1) (9,10,11). Of note, many of the known risk factors are particularly germane for children; for example, premature neonates are at the extremes of age, often receive polypharmacy, and have developmental impairments in renal and hepatic drug clearance (12). Another underestimated factor with respect to developmental pharmacology is that the enhanced capacity of some enzymatic pathways in toddlers may also place them at increased risk for activation-induced ADRs (13). Children also cannot evaluate and express their response to medication as well as their adult counterparts.
Classification of Adverse Drug Reactions
A number of systems have been proposed to classify ADRs. At the onset, it is important to acknowledge a major nosological issue with respect to popular designation of adverse events associated with therapy. There is a common and regrettable use of the word “allergy” to apply as a blanket descriptor for all adverse events that occur in temporal relationship to drug therapy. This is both inaccurate and misleading. Although allergy in popular usage is considered to be sensitivity to a drug or chemical, in fact allergy is a very specific term, which a purist might define as an immunoglobulin E–mediated adverse event, but which is more commonly considered as an adverse reaction to a drug mediated by the immune system (14). The latter definition is much more useful, as it both speaks to mechanism and helps to guide clinical decision making as to diagnosis and therapy.
The most useful system for classifying ADRs is that described by Rawlins and Thomas, who in 1977 proposed that ADRs should be considered to be predictable or unpredictable (Table 59.2) (15). Predictable ADRs are
very common and typically not very severe. In contrast, unpredictable ADRs are much less common but often much more severe. As well, given their relatively uncommon occurrence, the vast majority of unpredictable ADRs are not described until the drug in question has gone through the development process and has been approved for general use. To illustrate, the majority of drugs are evaluated in 5,000 or fewer patients under tightly controlled circumstances prior to marketing. Many serious drug hypersensitivities occur at an incidence of 1:1,000 to 1:5,000 patients, an incidence that has potential impacts on public policy and drug regulation. However, there is essentially no chance that a hypersensitivity reaction that occurs at this rate will be detected prior to drug approval and entry into the market. This has significant implications in that serious hypersensitivity information would therefore become part of the product monograph either late or never.
very common and typically not very severe. In contrast, unpredictable ADRs are much less common but often much more severe. As well, given their relatively uncommon occurrence, the vast majority of unpredictable ADRs are not described until the drug in question has gone through the development process and has been approved for general use. To illustrate, the majority of drugs are evaluated in 5,000 or fewer patients under tightly controlled circumstances prior to marketing. Many serious drug hypersensitivities occur at an incidence of 1:1,000 to 1:5,000 patients, an incidence that has potential impacts on public policy and drug regulation. However, there is essentially no chance that a hypersensitivity reaction that occurs at this rate will be detected prior to drug approval and entry into the market. This has significant implications in that serious hypersensitivity information would therefore become part of the product monograph either late or never.
Table 59.1 Risk Factors for Adverse Drug Reactions | ||||||||
|---|---|---|---|---|---|---|---|---|
|
Table 59.2 Classification of Adverse Drug Reactions | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Mechanisms of Adverse Drug Reactions
As would be expected, the mechanisms of ADRs are dependent on the nature of the ADR, as outlined above. In the case of predictable ADRs, adverse consequences of therapy are the result of classical pharmacological determinants, and thus at different stages of development, children may be at special risk when ontologically determined alterations in pharmacokinetics or pharmacodynamics change how drugs are eliminated or how drugs act. As well, the circumstances under which drug therapy is decided upon and under which drugs are administered may predispose children at certain times in life to an increased risk for drug toxicity (Fig. 59.1). These will be considered in turn.
Developmental Pharmacology
A unique issue with respect to ADRs in children, notably preterm infants, is developmental pharmacology (12). Although the admonition that “children are not small adults” is true across childhood, nowhere is this more important than in the first year of life, especially among infants born preterm. In terms of the classical pharmacological rubric of Absorption, Distribution, Metabolism, and Excretion, variations exist across all four domains, with potential impacts on the risk for ADRs.
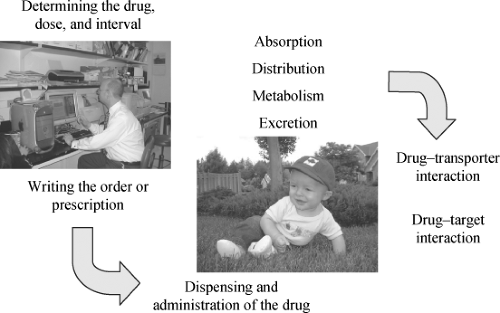 Figure 59.1. A number of factors are interrelated with respect to risk for adverse drug reactions in children, including determining the drug choice, writing the drug dose and dose interval order, dispensing and administering the drug, as well as pharmacokinetic and pharmacodynamic factors: drug absorption, distribution, metabolism, excretion, and drug action. |
Absorption
The vast majority of drugs used in the therapy of children are given by the oral route, and there are well-described developmental differences in oral and gastrointestinal physiology between infants and older children and adults (16,17). These changes include the near-neutral pH of the newborn’s stomach compared with the acidic stomach pH seen in adults and older children, delayed gastric emptying seen for the first 6 months of life, variable intestinal transit time, immature intestinal function, and altered capacity for both intestinal wall metabolism and transport. Many of these changes, notably with respect to metabolism and transporter function, remain poorly understood. The overall impact of these changes on the risk for ADRs is, for most drugs, minor. However, for some drugs, such as phenytoin, the marked differences between absorption in infants and older children place infants at an increased risk for both therapeutic failure and ADRs. Drug absorption when drugs are given via the intramuscular and pulmonary routes can vary greatly in infants, related to diminished muscular mass and muscular blood flow and to differences in pulmonary function between infants and adults (18). As well, the skin of newborns demonstrates transient, but significant, increase in percutaneous drug absorption compared with the skin of older infants, who in turn have enhanced percutaneous drug absorption compared with adults (19). This can be associated with toxicity for drugs that are not normally toxic by the cutaneous route in adults, especially if administered in large amounts (20).
Distribution
There are substantial differences in body composition between infants and adults, with infants having 80% body water compared with 55% in adults and extracellular water
of 45% in infants compared with 20% in adults (17). Also, plasma protein binding in infants is less extensive than in adults, which appears to be due to a mixture of relatively reduced binding capacity as well as relatively reduced binding affinity. As well, the blood–brain barrier of the infant is less well developed than that of adults (17). Thus, drugs that are highly protein bound may have a somewhat larger volume of distribution in infants than in adults, while drugs that normally do not cross the blood–brain barrier may be able to do so in infants. That being said, the impact of these changes on risk for ADRs is, overall, quite small.
of 45% in infants compared with 20% in adults (17). Also, plasma protein binding in infants is less extensive than in adults, which appears to be due to a mixture of relatively reduced binding capacity as well as relatively reduced binding affinity. As well, the blood–brain barrier of the infant is less well developed than that of adults (17). Thus, drugs that are highly protein bound may have a somewhat larger volume of distribution in infants than in adults, while drugs that normally do not cross the blood–brain barrier may be able to do so in infants. That being said, the impact of these changes on risk for ADRs is, overall, quite small.
Metabolism
It is now well recognized that infants, notably preterm infants, have substantial differences in metabolic capacity and metabolic pathways when compared with older children and adults (12). This difference in metabolism has translated into therapeutic disaster on numerous occasions. One of the first, and among the most notable, was the chloramphenicol Grey baby syndrome (2,21,22). In the late 1950s, a syndrome characterized by cardiovascular instability with rapid progression to death was noted among infants being treated with chloramphenicol for possible sepsis (21). It has subsequently been appreciated that these infants had developmental limitations in the rate at which chloramphenicol could be metabolized via glucuronidation, with the consequence of immature glucuronidation being accumulation of chloramphenicol producing dose-dependent mitochondrial toxicity (22). This tragedy illustrated how developmental immaturity in drug metabolism capacity places neonates—especially premature neonates—at special risk (23).
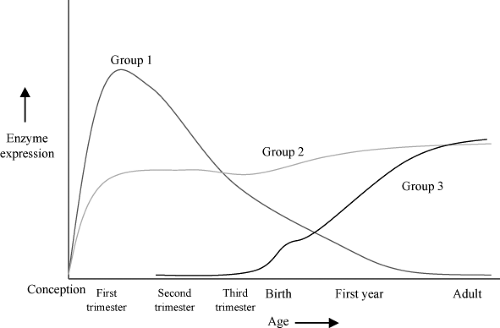 Figure 59.2. A schematic representation of the three major patterns of enzyme ontogeny (Hines RN. The ontogeny of drug metabolism enzymes and implications for adverse drug events. Pharmacol Ther 2008;118:250–267): with Group 1 being enzymes that are primarily expressed during the first and second trimester, Group 2 being those enzymes that are expressed relatively constantly during gestation, and Group 3 being those enzymes whose expression increases dramatically after birth. Examples of Group 1 enzymes include CYP3A7, FMO1, SULT1A3/4, and SULT1E1. Examples of Group 2 enzymes include CYP2C19, CYP3A5, and SULT1A1. Examples of Group 3 enzymes include CYP1A2, CYP2C9, CYP2D6, CYP2E1, CYP3A4, FMO3, ADH1B, ADH1C, and SULT2A1.
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|