Computed tomography angiogram of a young woman with Marfan syndrome. The aortic valve/root and ascending aorta have already been replaced, as has much of the descending thoracic aorta. A transvenous pacemaker prevented magnetic resonance imaging being used for routine follow-up imaging. The abdominal aorta has two extensive areas of aneurysmal dilatation extending down to the iliac arteries.
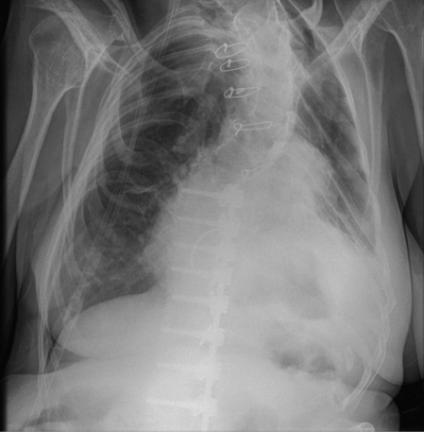
Plain chest X-ray of a patient with Marfan syndrome. This shows significant scoliosis associated with a previous Harrington rod operation. There is evidence of previous cardiac surgery—this included replacement of the aortic and mitral valves, the aortic root, and the ascending aorta. The patient also has a marked restrictive lung defect secondary to Marfan syndrome (oxygen tubing for regular oxygen supplementation can be seen).
Only a proportion of Marfan patients will be fully aware of their diagnosis and under appropriate specialist follow-up. Knowledge of the implications of pregnancy and the opportunity for prepregnancy counseling is not universal. This is a concern because important health-checks may be omitted in an asymptomatic mother and the opportunity for informed decision-making lost.
Nonaortopathy complications of Marfan syndrome
Aortic aneurysm formation and dissection are, by far, the most concerning features of Marfan syndrome. However, Marfan syndrome is a multisystem connective tissue disorder with other sequelae that may have an impact on all stages of pregnancy and delivery.
Mitral valve regurgitation due to a floppy or prolapsing mitral valve is another common cardiac manifestation.[14] Regurgitant lesions are usually tolerated well during pregnancy. An exception to this would be if there was already a degree of ventricular dysfunction that would be adversely affected by further volume loading. A subset of Marfan patients does have intrinsic ventricular impairment and this is often unmasked during pregnancy, with further left ventricular dilatation and symptomatic ventricular ectopy.
A number of obstetric complications have been described in patients with Marfan syndrome (Table 12.2). Whether these are causal or chance associations is unclear.
| Aortic dissection, aneurysm formationPost |
| Aortic regurgitation |
| Left ventricular dysfunction |
| Mitral valve prolapse ± regurgitation |
| Early pregnancy loss—possible association with habitual abortion |
| Premature rupture of membranes |
| Pelvis instability |
| Chronic back pain |
| Possible cervical incompetence and uterine inversion |
| Postpartum hemorrhage |
| Poor fetal outcome (including 50% recurrence risk) |
Pregnancy as a risk factor for aortic disease
A relationship between aortic dissection (from all causes) and pregnancy was first reported in 1944.[15] However, the validity and strength of this relationship is debatable and the published literature may be misleading. Publication bias and the plethora of case reports would suggest a strong relationship. Indeed the earliest papers suggested than in Marfan patients under the age of 40 years, 50% of dissections were associated with pregnancy.[16,17]
In contrast, recent cohorts have suggested that the association is less extreme.[18] Meijboom et al. reported in 2005 a follow-up study of 127 Marfan patients and was able to study prospectively 33 pregnancies in 23 women.[19] They documented remarkably stable aortic root diameters throughout pregnancy and the puerperium. Only one woman with a previous type A dissection developed an aortic complication (type B dissection) during her second pregnancy. They stated that in patients with no history of aortic involvement, dissection during pregnancy is uncommon and that “Pregnancy in women with Marfan syndrome seems to be relatively safe up to an aortic root diameter of 45 mm.”
Risk stratification
The European Society of Cardiology and the American Heart Association/American College of Cardiology have produced guidelines on the optimal management of Marfan patients considering pregnancy (Table 12.3).[20,21] In addition, Marfan syndrome self-help groups also distribute written and electronic information to their members. There are some differences between these guidelines but they agree on the general principles governing management.
| Guideline | Aortic root dimensions | Medical therapy | Delivery |
|---|---|---|---|
ESC Pregnancy Guideline (2011) [20] | If >45 mm, surgery should be performed prepregnancy | Beta-blockers | Consider cesarean if aorta >45 mm |
| Patients with type B dissection should be advised against pregnancy | |||
| ACCF/AHA Aortic Guideline (2010) [21] | If >40 mm surgery is reasonable prepregnancy | Beta-blockers | Consider cesarean for “significant aortic dilatation” |
ACCF, American College of Cardiology Foundation; AHA, American Heart Association; ESC, European Society for Cardiology
Although the majority of dissections occur due to proximal root dilatation, fatal dissection can occur outwith this setting. Dissection can occur in “low-risk” aortas that are not dilated.[22] Although rare, the patient needs to be aware that a reassuring echocardiogram does not equate to no risk. Family history and rate of change in aortic dimensions are other important components of quantifying risk.[23] Distal aortic dissection (type B dissection) is less predictable. Immer et al. in 2003 suggested that for proximal dissections root diameter and rate of change were reliable predictors but that there were no equivalent predictors for the distal aorta.[24] The distal aorta also has the disadvantage of being more difficult to image than the aortic root because of very limited echocardiography windows.
Successful pregnancies have been documented in patients with previous aortic root replacement but most centers still regard these patients as being at a significant risk of further events. When a composite metal valve and graft have been used, the risks associated with anticoagulation further complicate care.[25]
It should be reiterated that the absence of risk factors does not equate with no risk. In comparison with the average health woman in developed countries (maternal mortality <1 in 10 000 in the UK), any Marfan pregnancy is “high risk.”
Medical therapy
Chronic beta-blockade, originally with high-dose propranolol, appeared to show a slowing of the rate of aortic root enlargement in patients with Marfan syndrome.[26] This was thought to be due to direct blood pressure effects, changes in aortic compliance, and alterations in aortic pressure–area relationships.[27] More recent meta-analysis has questioned the validity of this effect.[28] During pregnancy, the risk vs benefit of any therapy needs to be discussed fully; however, despite the association of beta-blockers with fetal growth restriction, for the majority of patients it is advised that they should be continued. Newer agents, such as angiotensin receptor blockers, may potentially have a more profound impact on aortic dilatation.[29] These agents are, however, contraindicated in pregnancy (see Chapter 7).
Recurrence risk
Most familial or genetic aortopathies have an autosomal dominant inheritance. This means a 50% recurrence risk. For many individuals with Marfan, this is an unacceptably high risk. These individuals may, in the past, have chosen not to have children. More recently, both prenatal diagnosis and preimplantation genetic testing techniques have become possible if the family mutation is known. Prenatal diagnosis using chorionic villus sampling can be performed at 13 weeks, with the option of terminating an affected pregnancy. For those not wishing to go through a termination of pregnancy, preimplantation genetic testing using in vitro fertilization techniques is an alternative. These options are only available when a causal mutation in the fibrillin gene has been identified (there are a number of different mutations, and only about 80% of women with Marfan syndrome will have one that is currently identifiable).[30] Patients should be referred to a clinical geneticist early because genotyping and antenatal counseling may be a prolonged process.
Other autosomal dominant aortopathies
As genotyping improves, a new group of familial genetic aortopathies are being diagnosed. For example mutations in the ACTA2 gene leads to aortopathy and dissection. There has been a small series of 137 delivered pregnancies with maternal ACTA2 mutation reported: there were eight dissections (six type A and two type B) and one coronary dissection.[31] Another well-recognized aortopathy is Loeys-Dietz syndrome. This condition can be secondary to mutations in several different genes, including TGFBR1, TGFBR2, SMAD3, and TGFB2. Again, multisystem involvement is common, but there have been few reported pregnancies in this cohort to date.
Monitoring during pregnancy
Echocardiography is the foundation of aortic imaging for the well patient. This can simply and noninvasively track the diameters of the aortic root and assess the aortic valve. The aortic root is best measured at four levels (the “annulus,” the sinuses of Valsalva, the sinotubular junction, and in the ascending aorta) from the long-axis parasternal window. Imaging every 4–6 weeks has been suggested by several groups.[32]
Magnetic resonance imaging (MRI) can be performed during pregnancy. On a “safety first” basis, some clinicians recommend only scanning after the first trimester, although there are no reported hazards of MRI to the fetus. MRI is important to detect any areas of aortic dilatation that are difficult to see with ultrasound—this is particularly important in non-Marfan aortopathies, where the pattern of arterial involvement is different. Transesophageal echocardiography (TOE) is an alternative method of imaging the thoracic aorta and root but is less helpful in imaging the arch. During pregnancy, the increased risk of gastroesophageal reflux means that TOE should be done with a protected airway (i.e. under a general anesthetic with endotracheal intubation). In practice, TOE is rarely needed. Computed tomography gives a significant dose of radiation exposure and should only be used when there is no alternative technique and when the benefit of the information obtained outweighs this risk.
Symptoms of dissection
Catastrophic cardiovascular collapse may be the first sign of dissection, in which case the diagnosis is often clear. Other symptoms heralding a new event include chest and back pain, dizziness, collapse, and haemoptysis. Caution should be exercised with the patient who presents with suspected pulmonary thromboembolism and has negative investigations. Patients should be warned regarding symptoms of dissection and encouraged to actively request cardiac review if they present unwell to their local hospital. A low threshold for urgent imaging is essential.
Treatment of aortic dissection or “near dissection” during pregnancy
If echocardiography documents rapidly increasing aortic root diameters during pregnancy, then difficult management decisions need to be faced. New aortic regurgitation may also be another indication of a developing “unstable” situation.
If the fetus is viable, it has been suggested that the patient should be delivered promptly and then undergo urgent, or semiurgent, aortic root surgery.[33] This is usually either a composite root and valve graft, or a valve–sparing aortic root replacement retaining the patient’s own aortic valve. If the clinical situation allows, there should be a window between delivery and subsequent cardiac surgery (some units suggest 48 h) to minimize the risks of postpartum bleeding.
If pregnancy is at an earlier stage, root surgery may need to be undertaken with the fetus in situ. There are cardiopulmonary bypass techniques that can minimize fetal risk. Even so, bypass is associated with fetal loss.[34] It is hoped that normotensive high-flow, high-pressure techniques will minimize the hypotensive ischemic insult to the fetoplacental bed.[35]
It may be possible to treat distal dissections nonsurgically—assuming there is no extravasation of blood and no loss of arterial supply to a critical organ. Endovascular repairs are possible, but there are no data regarding this technique during pregnancy. As with all dissections, meticulous attention to blood pressure is paramount.
Delivery issues
Obstetricians and cardiologists hold strong views regarding the optimal way to deliver mothers with cardiac disease. However, outwith the setting of breech delivery there is no randomized data to suggest whether vaginal delivery or cesarean section is systematically superior.
In this population the important principles determining delivery are:
1. Timing—when there is evidence of an unstable aorta, delivery may need to be before term and at short notice
2. Adequate control of blood pressure with invasive monitoring
3. Minimization of the catecholamine response and resulting aortic sheer stress.
If the woman is having a vaginal delivery, this usually means a fully assisted second stage with regional analgesia. Marfan patients often have dural ectasia and spinal deformities. This potentially increases the likelihood of a failed or patchy epidural,[36] although in practice this is rarely a clinical problem. A previous spinal rod is usually a contraindication to a regional anesthetic because of the concerns regarding infection.
The possibility of postpartum hemorrhage should be anticipated; however, bolus doses of uterotonic agents, such as ergometrine (can cause a rapid rise in blood pressure) or oxytocin (can cause a sudden fall in blood pressure) should be avoided. In the presence of a prosthetic valve, prophylaxis against endocarditis should be considered for cesarean or instrumental deliveries, although this remains controversial.
The puerperium is a time of particular high risk for the aortopathy patient. During this period, meticulous blood pressure control and further cardiac assessment should be performed. Patients with aortopathy are not appropriate for early discharge and should be monitored in hospital until the majority of the cardiovascular changes following delivery have been observed—this usually takes 3 to 5 days.
Coarctation of the aorta
Coarctation of the aorta (a narrowing in the aorta, usually at the site of the duct) accounts for 7% of congenital heart disease. Although the majority of cases present neonatally, coarctation can be diagnosed at any age and may first present during pregnancy. Coarctation is commonly associated with other congenital and acquired cardiovascular abnormalities which may impact pregnancy (Table 12.4). This section will focus on simple (isolated) coarctation.
| Associated lesions |
| Bicuspid aortic valve (± regurgitation ± stenosis) |
| Proximal aortopathy |
| Patent duct—usually closed neonatally |
| Ventricular septal defect |
| Hypoplastic aortic arch |
| Abnormal head and neck vessels (5%) |
| Intracerebral berry aneurysms (5%) |
| Multilevel left heart obstruction (e.g. subaortic stenosis, aortic valve disease, parachute mitral valve = Shone syndrome) |
| Hypoplastic left heart syndrome |
| Associated sequelae |
| Hypertension |
| Aortic obstruction (native or recoarctation) |
| Lower body hypotension (effecting placenta) |
| Para-coarctation aneurysm |
| Aortic rupture (especially if patch repair or bicuspid valve) |
| Left ventricular hypertrophy ± failure |
| Late atherosclerotic disease (coronaries, cerebral) |
| Endocarditis |
In cardiac obstetric practice, the majority of mothers with coarctation will have had previous surgical intervention (in the series described by Beauchesne et al., about 70%[37]). However, even following successful surgical repair, important sequelae remain. The most significant of these are hypertension, recoarctation, and aneurysm formation.
Native coarctation
Patients with undiagnosed coarctation may present during pregnancy. This may be the first occasion that the patient has had a clinical examination or an assessment of their blood pressure since childhood. Significant hypertension, especially systolic hypertension, in the first trimester should alert medical staff to the possibility of an occult coarctation. A systolic murmur, especially over the left scapula, may also be present. Good practice in women with hypertension should include a measurement of the blood pressure in both arms and an assessment of leg pulses (and/or leg blood pressures).
Diagnosis can often be confirmed with echocardiography, although when requesting this examination, specific mention of the possibility of a coarctation should be made to ensure that appropriate images of the aortic arch are obtained. Other imaging modalities (such as MRI) should also be used to assess any areas of aortic dilatation, detect the presence of collateral arteries, and plan intervention if required.[38] It is not appropriate to rely on an ultrasound examination alone to conclude that a woman does not have a coarctation if this is considered clinically to be a possibility (e.g. a marked discrepancy between the blood pressure in the arms and the legs—high in the arms, and low in the legs).
The presence of a significant coarctation is potentially difficult to manage during pregnancy and, indeed, both the mother and the fetus are at risk. The major concern is severe maternal hypertension that is refractory to drug therapy and, paradoxically, hypotension of the fetoplacental unit (although the blood pressure in the upper body is often high, the blood pressure below the coarctation may be markedly reduced, thus limiting the blood flow to the placenta). When severe coarctation is detected the options include attempting to progress with medical therapy (antihypertensives), surgical repair of the coarctation with the fetus in situ, attempting transcatheter coarctation stenting, or terminating the pregnancy. Expert multidisciplinary input is needed to make these complex management decisions.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


