Chapter 47
Malignant Hyperthermia
M. Joanne Douglas MD, FRCPC
Chapter Outline
PREGNANCY AND MALIGNANT HYPERTHERMIA
MANAGEMENT OF THE MALIGNANT HYPERTHERMIA–SUSCEPTIBLE PARTURIENT
Obstetric Drugs in Malignant Hyperthermia–Susceptible Parturients
ASSESSMENT OF HYPERTHERMIA AND TACHYCARDIA
Malignant hyperthermia (MH) is an inherited disorder of skeletal muscle. On exposure to triggering agents (e.g., succinylcholine, volatile halogenated anesthetic agents), affected individuals demonstrate a hypermetabolic syndrome characterized by hypercapnia, acidosis, muscle rigidity, arrhythmias, and hyperthermia. MH was first described in 1960 by Denborough and Lovell,1 but may have been responsible for some of the earlier deaths attributed to ether and chloroform anesthesia.2
Epidemiology
Ørding,3 reviewing the incidence of MH in Denmark, noted that the incidence of the fulminant syndrome (e.g., muscle rigidity, acidosis, hyperkalemia, arrhythmias, hyperthermia, increased creatine kinase [CK] levels, myoglobinuria) was 1 in 220,000 patients who received general anesthesia and 1 in 62,000 patients in whom succinylcholine was combined with a volatile halogenated agent. MH (either mild or fulminant) was suspected in 1 in 16,000 patients who received anesthesia of any type. The male-to-female ratio was 1.4 : 1.3 There is some geographic variation in the incidence of MH.4
There are few reports of development of MH during pregnancy and parturition.5–12 The infrequent occurrence during pregnancy probably reflects both the low frequency of this disorder in the general population and the widespread use of local and neuraxial anesthetic techniques in obstetric patients.
Pathophysiology
MH is the result of a disorder in the regulation of intracellular calcium in skeletal muscle. The precise mechanism by which volatile anesthetics and depolarizing muscle relaxants cause an MH crisis is still unknown.13 In muscle the sarcoplasmic reticulum is responsible for controlling calcium release and reuptake during muscle contraction.14 During skeletal muscle excitation-contraction coupling, calcium is released from the terminal sarcoplasmic reticulum via the ryanodine receptor (RYR1). Dihydropyridine receptors in the T-tubule membrane also participate in the excitation-contraction coupling. In humans, mutations in both the dihydropyridine receptor and the ryanodine receptor can result in clinical MH. Dantrolene inhibits excitation-contraction coupling, and succinylcholine, caffeine, and volatile halogenated agents increase it.
Genetics
MH is a heterogeneous disorder, meaning that more than one gene defect is responsible for expression of the clinical syndrome.15 It is inherited in an autosomal dominant fashion with variable penetrance, although this pattern has been questioned in some families.16 Porcine MH is transmitted as a recessive gene. The defective gene in MH-susceptible pigs has been localized to a single point mutation in the ryanodine receptor gene (RYR1) responsible for the calcium release channel.17
Investigators have found the corresponding point mutation on the human ryanodine receptor gene (RYR1) in some families with MH (chromosome 19q12.1-13.2; locus MHS-1), and other mutations in RYR1 have been linked to MH susceptibility.18 Other point mutations in RYR1 are found in patients with central core disease, a myopathy associated with MH.19 As of 2011, more than 200 RYR1 mutations associated with MH had been identified.15 Mutations responsible for MH in some families are located on chromosomes 5p, 17, 7q, 3q, and 1q.20 Other myopathies may be characterized by a hyperthermic state with muscle damage and metabolic derangements similar to those seen in MH, but their chromosomal abnormality has not been mapped to the same area.15
Triggers
Known triggers of MH include the depolarizing muscle relaxants (e.g., succinylcholine) and all the volatile halogenated anesthetic agents (i.e., sevoflurane, desflurane, isoflurane, halothane, enflurane) (Box 47-1). The dose and duration of exposure to the triggering agent may influence the onset and severity of a reaction. Previous uneventful administration of general anesthesia with triggering anesthetic agents does not rule out the diagnosis of MH.21
In contrast to the porcine model, reports of stress-induced MH in humans are rare.22–28 In one report two cases of fatal, stress-induced MH occurred in unrelated families26; both children had a known RYR1 mutation and one had a second mutation, possibly suggesting an additive effect.29 The sympathetic nervous system is active during an episode of acute MH, but there is insufficient evidence to implicate increased sympathetic activity as a cause in humans. Although muscle biopsy testing may help distinguish MH from exercise-induced myolysis, exertional heat stroke, and other myopathies, there is some evidence of a link between some cases of heat stroke and exercise-induced rhabdomyolysis and MH susceptibility.30
Investigators have explored other possible triggers of MH both in the porcine model and in humans. No evidence suggests that exogenous calcium, digoxin, hypercarbia, potassium,31 or norepinephrine32 triggers MH. Exercise33 and environmental temperature34–36 may intensify an existing reaction or modify a developing reaction. Sodium thiopental and pancuronium delay the onset in pigs and may modify the reaction in humans.37 Duke et al.38 postulated that hypomagnesemia may increase the probability and severity of an MH event in MH-susceptible humans.
There are case reports of the occurrence of MH during regional anesthesia and during general anesthesia with nontriggering agents.39–43 The cases that occurred during regional anesthesia appeared mild and responded readily to treatment. In some cases, however, the diagnosis was not confirmed with muscle biopsy or appropriate laboratory investigation at the time of the event.
Clinical Presentation
Individuals who are MH-susceptible may demonstrate the fulminant syndrome when anesthetized with a triggering agent. During an acute episode, the diagnosis is based on the finding of an elevated end-tidal CO2 concentration, muscle rigidity (generalized and/or masseter), respiratory and metabolic acidosis, rhabdomyolysis, hyperkalemia, elevated CK concentration, and myoglobinuria (Box 47-2). Hypoxemia, unstable blood pressure, and evidence of sympathetic hyperactivity (e.g., tachycardia, hypertension, arrhythmias) are other signs. Hyperthermia may occur early, but often it is a late sign. Perioperative rhabdomyolysis, without any of the previously mentioned clinical signs, also may indicate MH susceptibility.44,45
With the advent of routine end-tidal CO2 monitoring, MH may be detected early, often before the development of rhabdomyolysis and hyperthermia.46 This situation may lead to uncertainty about the clinical diagnosis of MH, given that many of the confirmatory signs and laboratory abnormalities may be absent during the early phase of MH. Thus, early treatment of possible MH could present a dilemma as to whether the patient should undergo diagnostic muscle biopsy or should be assumed to be MH susceptible.
Masseter Muscle Rigidity
Masseter muscle rigidity is one of the early signs of MH.46 The masseter muscles are sensitive to the action of succinylcholine and respond with increased tension in normal individuals.47,48 Often this tension is imperceptible, but in some patients it is impossible to open the mouth for laryngoscopy and intubation. The duration of rigidity parallels the duration of action of succinylcholine. Typically there is no difficulty with mask ventilation. Masseter muscle rigidity rarely occurs after the use of nondepolarizing muscle relaxants such as rocuronium and vecuronium.49 Patients with myopathies and other neuromuscular disorders also may have masseter muscle rigidity after the administration of succinylcholine.50
If masseter muscle rigidity is accompanied by generalized rigidity, anesthesia should be discontinued, dantrolene should be administered, and the patient should be monitored closely.51 However, there is controversy regarding the management of isolated masseter muscle rigidity.52 Options include (1) discontinuation of the anesthetic agents and administration of dantrolene; (2) continuation of anesthesia with nontriggering, “safe” agents and close attention to the end-tidal CO2 concentration; and (3) continuation of anesthesia with triggering agents and careful monitoring. In my judgment, the anesthesia provider should either discontinue anesthesia altogether or continue anesthesia with nontriggering agents. If the anesthesia is continued, the minute ventilation, end-tidal CO2 concentration, electrocardiogram (ECG), temperature, and arterial blood gas measurements should be monitored. The anesthesia provider also should look for evidence of rhabdomyolysis by monitoring CK levels and looking for myoglobinuria, and should recommend that the patient undergo muscle biopsy and caffeine-halothane contracture test (see later discussion).53
Diagnosis
Investigators have correlated clinical presentation (i.e., evidence of metabolic and muscle derangements) with abnormal contracture on the caffeine-halothane contracture test.54–56 The greater the number of clinical signs or abnormal laboratory findings, the greater the risk for MH (Table 47-1).54 An early assessment of the risk for MH allows the anesthesia provider to initiate appropriate treatment. The mortality rate for MH is as high as 80% without dantrolene therapy.56 Early administration of dantrolene lowers the mortality rate to 4%.57
TABLE 47-1
Risk for Malignant Hyperthermia (MH) with Associated Signs and Symptoms
| Type | Symptoms/Signs | Risk |
| Fulminant/classic | Metabolic acidosis | 0.96 |
| Muscle rigidity | ||
| Hyperthermia (> 38.5° C) | ||
| Arrhythmias | ||
| Hyperkalemia | ||
| Myoglobinuria | ||
| Increased creatine kinase level | ||
| Moderate | Inconclusive signs of MH involving metabolic and muscle abnormalities, with MH the probable diagnosis | 0.88 |
| Mild | Signs of metabolic derangement (pH > 7.3, body core temperature < 38.5° C) | 0.14 |
| Masseter spasm with rhabdomyolysis | Creatine kinase level > 1500 U/L, myoglobinuria | 0.76 |
| Masseter spasm with signs of metabolic disturbance | Arrhythmias, rising core temperature | 0.57 |
| Masseter spasm only | 0.28 | |
| Unexplained perioperative death or cardiac arrest | 0.66 | |
| Other | Postoperative pyrexia or rhabdomyolysis | 0.07 |
Data from Ellis FR, Halsall PJ, Christian AS. Clinical presentation of suspected malignant hyperthermia during anaesthesia in 402 probands. Anaesthesia 1990; 45:838-41.
An international group of experts has developed a clinical grading scale to predict MH susceptibility.58 This scale consists of six processes (rigidity, muscle breakdown, respiratory acidosis, temperature increase, cardiac involvement, family history) and their clinical indicators. Points are assigned for each indicator present in a patient, and the total represents a raw score. A rank is subsequently assigned to this score, which indicates the likelihood of development of MH in the patient.
Testing
Susceptibility to MH is determined by a positive caffeine-halothane contracture test result. During this test, fresh muscle is exposed to halothane and caffeine, and the extent of contraction is measured. The caffeine-halothane contracture test has been standardized in MH testing centers throughout North America (the North American protocol)59 and Europe (the European protocol is called the in vitro contracture test).60 This test is the “gold standard” for the diagnosis of MH. The sensitivity and specificity of the North American protocol are 97% and 78%, respectively.61 Some false-positive results may occur.62 Patients with a negative caffeine-halothane contracture test result subsequently have received anesthesia with triggering agents without incident.63–66
Testing for the known genetic mutations associated with MH is now available. However, because all the genetic mutations responsible for MH have yet to be identified, genetic testing is still not sensitive enough to use for routine screening.67 In the future, MH may be detected in most MH-susceptible patients without the need for an invasive muscle biopsy.67 In the absence of muscle biopsy results, a parturient with a positive family history should be treated as if she were MH susceptible.
Pregnancy and Malignant Hyperthermia
In 1972, Crawford68 wondered “whether or not there was a record of a pregnant or newly born patient or animal having developed hyperpyrexia and … whether hyperpyrexia has been encountered in a patient undergoing an operation under regional block anesthesia.” Subsequently there have been few reports of MH during parturition and fewer reports of maternal mortality attributable to MH. Wadhwa5 reported the death of a woman with a known family history of MH in whom muscle rigidity developed during twilight sleep for parturition. Douglas et al.6 reported one fatal case of MH in a parturient undergoing general anesthesia for cesarean delivery.
There are three published reports7–9 of nonfatal MH during cesarean delivery and one report of MH after cesarean hysterectomy performed because of postpartum hemorrhage.12 The triggering agents were succinylcholine and halothane,7 succinylcholine and isoflurane,12 cyclopropane,8 and succinylcholine alone (without a volatile halogenated agent).9 There are several reports of the successful administration of epidural and spinal anesthesia during labor and cesarean delivery in MH-susceptible parturients.5,12,69–76
Although it is unclear whether pregnancy alters susceptibility to MH, the rarity of MH events during pregnancy suggests that pregnancy protects against the occurrence of MH. However, it also may reflect the widespread use of neuraxial anesthesia for labor, vaginal delivery, and cesarean delivery.
Maternal Physiology
Basal metabolic rate, oxygen consumption, and minute ventilation increase during pregnancy (see Chapter 2). Serum bicarbonate, buffer base, and base excess decrease to maintain normal pH. Thus, the pregnant patient typically has a compensated respiratory alkalosis. The reduced buffering capacity could adversely affect the pregnant woman during an episode of MH.
Oxygen consumption and minute ventilation increase further during labor. Maternal lactate and pyruvate concentrations increase steadily during labor, indicating an increase in both aerobic and anaerobic metabolism. Hyperventilation during contractions may result in periods of hypoventilation between contractions, which may adversely affect the PaO2 of both the mother and the fetus. These metabolic and physiologic responses to pain are similar to the metabolic and physiologic changes that are observed during acute MH. Effective epidural analgesia decreases oxygen consumption and minute ventilation. If tachycardia and hyperventilation occur despite effective analgesia, they are more likely to signal an episode of MH.
Aortocaval compression from the pregnant uterus results in decreased cardiac output, hypotension, and reduced uteroplacental perfusion. Thus, aortocaval compression may accelerate the occurrence of acidosis during an episode of MH. Aortocaval compression hinders resuscitative efforts during cardiac arrest, and evacuation of the uterus (i.e., delivery of the fetus) facilitates maternal resuscitation.77 The obstetrician may need to deliver the fetus to facilitate maternal resuscitation during a fulminant case of MH.
CK concentrations are not diagnostic of MH. During pregnancy, there is a slight decrease in CK levels during the first trimester. CK levels remain stable until term, when they increase by approximately 50%. At delivery, there is an abrupt rise in the CK concentration, followed by a return to normal by 6 weeks postpartum (Figure 47-1).78 The increased CK concentration results from increases in both the CK-M fraction (skeletal muscle) and the CK-B fraction (myometrium, placenta, and fetal blood). Postpartum CK levels are higher in nulliparous women regardless of differences in duration of labor.79 Mean plasma CK activity is approximately 50% higher in African-Americans than in Caucasians or Asians.78
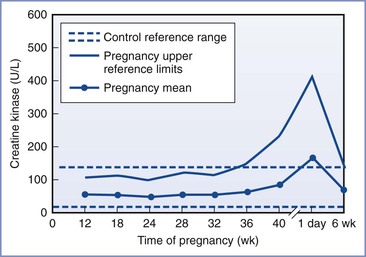
FIGURE 47-1 Changes in creatine kinase activity during and after pregnancy. (Modified from Lockitch G, editor. Handbook of Diagnostic Biochemistry and Hematology in Normal Pregnancy. Boca Raton, FL, CRC Press, 1993:59.)
Acute cocaine toxicity may mimic MH. Although cocaine does not induce contractures in MH-susceptible muscle,80 elevated CK concentrations and myoglobinemia can occur secondary to rhabdomyolysis and renal failure from cocaine intoxication.81
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


