Lymphomas
Lymphomas are a result of chromosomal alterations resulting in the uncontrolled growth of cells of lymphoid origin. Among all ages, lymphomas constitute just 5% of all cancers diagnosed annually in the USA. In children, however, this percentage increases to 8%.1 Combined, Hodgkin and non-Hodgkin lymphoma are the second most common childhood solid tumors (behind brain tumors and ahead of soft tissue sarcomas and neuroblastoma).
Lymphomas have classically been divided into two distinct groups: Hodgkin disease (HD) and non-Hodgkin lymphoma (NHL). In 2001, HD was designated Hodgkin lymphoma (HL) by the World Health Organization (WHO) lymphoma classification system.2 HL and NHL have a relatively similar prevalence among children and young adults, but NHL becomes significantly more common after 40 years of age (Fig. 69-1). Typically, patients with both HL and NHL are initially seen with enlarged lymph nodes and may have systemic symptoms of fever, fatigue and/or extralymphatic spread. However, these two types of lymphoma also have clear differences. HL usually is seen as an indolent process, whereas NHL is most often seen in children with a rapid onset of symptoms. Due to this propensity for rapid growth, children with NHL often have associated anatomic and metabolic co-morbidities to such a degree that their recognition and need for treatment constitutes a medical emergency. With HL, treatment is based primarily on staging and less on histologic subtype. In contrast, the current treatment of NHL depends on the histologic and immunophenotypic subtypes in addition to stage.
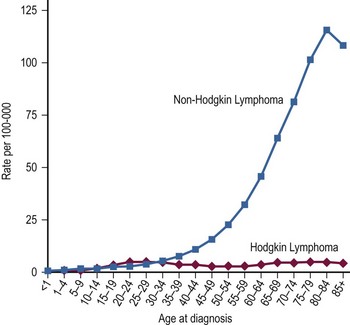
FIGURE 69-1 Age-specific incidence rates per 100,000 population for Hodgkin lymphoma and non-Hodgkin lymphoma from 2000–2009. (Howlader N, Noone AM, Krapcho M, et al, editors. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). Bethesda, MD: National Cancer Institute. http://seer.cancer.gov/csr/1975_2009_pops09/, based on November 2011 SEER data submission, posted to the SEER web site, 2012)
These two lymphomas are truly a study of contrasts. This is no more evident than in the evolution of their therapy. For years, HL has been one of the most curable cancers. Now, with markedly improved treatment protocols, NHL has a nearly equivalent cure rate.3 Children under 15 years of age had 5-year relative survival rates of 96% for HL and 86% for NHL from 2001–2007, up from 81% and 43%, respectively, from 1975–1977.1 Owing to the historic high survival rate with HL, its therapy has focused on a reduction in intensity. In contrast, because of its previously poor prognosis, NHL therapy has focused on intensification of therapy. The use of higher doses of chemotherapy over a short period (as compared with prior methods) has resulted in the dramatic improvement in cure and response of NHL.
Hodgkin Lymphoma
Thomas Hodgkin, in his classic thesis in 1832, described the gross necropsy examinations of seven patients.4 He noted the association of generalized lymphadenopathy and splenomegaly in six patients without evidence of infection or inflammation. Histologic descriptions of the Reed–Sternberg (RS) cell, the pathognomonic multinucleated giant cell, did not occur until after the turn of the century.5,6 Even though the etiology was unclear, therapeutic interventions began soon after the discovery of X-rays. More successful application of radiation therapy awaited the description of the disease’s propensity for contiguous spread. With this knowledge, application of radiation to the involved and adjacent nodal areas (extended-field technique) resulted in improvements in survival in the late 1930s.7 In the early 1960s, due to limitations in the radiologic techniques of that era, the practice of systematic laparotomy, splenectomy, and celiac node and liver biopsy at the time of initial presentation was developed for the purpose of staging and for targeted therapy.8 This has properly been described as the model for the careful staging of cancer as a required prerequisite to the design of therapy, which is a hallmark of oncologic practice today.9,10
During this same time, combination chemotherapy entered into the treatment armamentarium, and remission and cure rates markedly improved. These improvements have made HL one of the most curable cancers today, with a five-year survival of 96% for pediatric patients diagnosed between 2002 and 2008.11 With this high expectation for cure, attention over the past decade has focused on reducing the long-term sequelae of treatment. To this end, chemotherapy has evolved from an adjunctive role to a primary one, with the hope of eliminating the need for irradiation (and its attendant sequelae) altogether. When irradiation is needed, if used in combination with chemotherapy, the focus has been to reduce the size of the fields (from extended to involved) and the doses used. The two classic chemotherapy combinations (MOPP: nitrogen mustard, vincristine [Oncovin], procarbazine, prednisone; and ABVD: doxorubicin [Adriamycin], bleomycin, vinblastine, dacarbazine) have evolved. Hybrids of these combinations are now being utilized to reduce the toxicity to the patient.
Incidence and Epidemiology
It is estimated that 9,060 individuals will be diagnosed with HL in the USA each year, accounting for just 0.6% of all cancers and only 11% of all lymphomas.1 However, in children, it is the sixth most common type of cancer, with approximately 400 children diagnosed annually.1,12 This constitutes 4% of all childhood cases of cancer and approximately half of all childhood cases of lymphoma.1 HL has an incidence of 3.2 cases/100,000 teens aged 15–19 years.13 A bimodal distribution exists when considering all ages, but in children alone, a gradual trend is seen of increasing incidence with increasing age. HL is exceedingly rare in children younger than age 2 years and peaks in the adolescent years.14 Beyond age 11 years, it is the most common of the two types of lymphoma and accounts for about 15% of all cancer in young adults ages 15 to 24 years.13 A slight male predominance (1.3 : 1)11 is noted, but in the youngest children, the male-to-female ratio is much larger (12–19 : 1).15
Monozygotic twins of HL patients have been found to be at greater risk of developing HL than are dizygotic twins, strongly implicating genetics as a principal risk factor.16 In young adults, an increased risk of HL is found with higher socioeconomic status.17 Young adults with HL come from smaller families, have fewer infectious exposures as young children, and/or have later exposure to infections than do control populations.17,18 This correlates closely with socioeconomic status and implicates delayed exposure to infections as a principal risk factor.
Most likely, a combination of genetic risk and infectious exposure predisposes a young adult to HL. Immunodeficiency may be the link between these two risk factors, at least in a subgroup of HL patients. HL is more prevalent in human immunodeficiency virus (HIV)-infected patients.19–21 Also, patients with HL have a higher incidence of cellular immunodeficiency at the time of diagnosis.22 Etiologic theories encompass these two risk factors and focus primarily on the Epstein–Barr virus (EBV). Genomic material from EBV has been found in the RS cells in up to 79% of HL cases.23–25 A higher risk of HL has been noted in individuals with a history of infectious mononucleosis26–28 and with previously high titers to EBV.29 One hypothesis that incorporates these factors suggests the following sequence: (1) a genetic, iatrogenic, or viral immunosuppression; (2) subsequently or coincidentally, an EBV infection or oncogenetic rearrangement in a lymphoid precursor cell; (3) further genetic alterations; followed by (4) clonal expansion of lymphoid cells with morphologic features of RS cells; finally resulting in (5) the clinical syndrome known as HL, diagnosed by the presence of RS cells.30
Classification and Histologic Subtyping
The diagnosis of classical HL requires the dual finding of the diagnostic Hodgkin and RS cells (HRS cells) plus a reactive cellular background.31 The RS cell is a large cell (15–45 mm) with an ‘owl’s eye’ appearance (Fig. 69-2). It has a multilobed nucleus (or is multinucleated), each with a prominent eosinophilic nucleolus surrounded by a clear zone (halo) and an intensely stained nuclear membrane. The ‘owl’s eye’ appearance is the result of a bilobed nucleus. The RS cell often makes up no more than 2% of the involved cells. Hodgkin cells are the mononuclear variant of RS cells. The cellular background is a reactive, pleomorphic mixture of inflammatory cells including reactive lymphocytes, histiocytes, plasma cells, eosinophils, neutrophils, and fibroblasts, with varying degrees of fibrosis and sclerosis. The HRS cell is a clonal, neoplastic cell seen in classical HL and is thought to induce the reactive background through the abundant release of various cytokines.32 HRS cells typically are CD15 and CD30 positive and negative for CD45 and B-lineage antigens.33 In contrast, the nodular lymphocyte predominant (LP) HL cells (popcorn cells) are usually positive for B-lineage antigens, CD15 and CD30 expressions are lacking, and the immunoglobulin genes are expressed.33
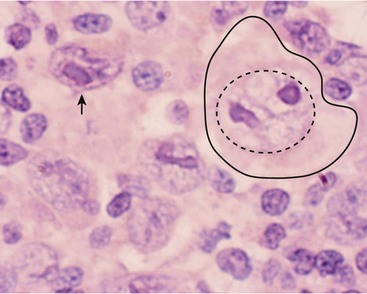
FIGURE 69-2 This photograph depicts a RS cell, which is pathognomonic for HD. On the right side of the slide, the large nucleolus is outlined by the dotted circle and the entire cell is outlined by the solid line. Note the relatively pale nuclear chromatin. The nucleolus has the appearance of an ‘owl’s eye’ from which it receives its name. The arrow points to a mononuclear variant of the RS cell, which has reticulated nuclear chromatin surrounding an almost rectangular macronucleus.
For histologic typing, the Rye classification was commonly used for three decades but has been supplanted by the WHO classification. The 2008 WHO classification lists two main types of HL: classical HL and nodular LP. Classical HL is further divided into four subtypes by morphology. These subtypes include nodular sclerosis (NS, the most common), mixed cellularity (MC), lymphocyte-rich (LRHL), and lymphocyte-depleted (LDHL).34 The NS subtype is seen in 40% of younger patients and 70% of adolescents.35 It is characterized by tumor nodules surrounded by broad sclerotic bands arising from a thickened fibrotic capsule.31 This subtype has a strong predilection for involving the lower cervical, supraclavicular, and mediastinal lymph nodes. The MC subtype is found in 30% of cases and has an increased incidence in younger children.15 HRS cells are typically increased in number. The lymph node architecture is often completely effaced by the HRS cells and their surrounding reactive cells. This subtype often is first seen with advanced, widely disseminated disease in extranodal sites. In addition to its relatively common incidence among all HL patients, it is the most common histologic type seen in HIV-infected patients.21
From 2001 to 2009, the National Cancer Institute (NCI)-sponsored SEER data revealed the following 5-year survival rates for patients age 0–19 years: LRHL 100%; NS 95.7%; MC 92%; and nodular LP 100% (Table 69-1). In patients of all ages, the 5-year survival rate for LDHL was 58%.11 Reports have shown that LPHL has a better prognosis and needs markedly reduced therapy to achieve cure.36,37 This differentiation of therapeutic response between LPHL and the other classic HL histologic types appears to validate the distinction observed in the immunophenotyped RS cells.38 The worse outcome of the MC and LDHL types may reflect their typically higher stage at diagnosis.
TABLE 69-1
Hodgkin Lymphoma: Sites of Involvement at the Time of Initial Diagnosis
aEleven per cent of patients were classified as NOS (not otherwise specified) and are not included in the histologic subtypes listed above.
Data from Howlader N, Noone AM, Krapcho M, et al, editors. SEER Cancer Statistics Review, 1975–2009 (Vintage 2009 Populations). Bethesda, MD: National Cancer Institute. http://seer.cancer.gov/csr/1975_2009_pops09/, based on November 2011 SEER data submission, posted to the SEER web site, 2012.
Clinical Presentation
Classically, children with HL present with painless enlarged lymph nodes, typically in the cervical or supraclavicular nodal groups (see Table 69-1). Nodes are often described as rubbery and fixed. They may be either single or matted with other nodes. Occasionally, because of rapid growth, tenderness may develop. Tumor lysis syndrome, a result of rapid and extensive tumor growth and a common complication in children with NHL, is rarely seen in children with HL.
HL tends to spread in a contiguous manner. Therefore, at presentation, one must examine carefully the nodal groups adjacent to the initially identified nodes. More than 90% of patients have involvement of either the cervical or mediastinal nodal groups (or both).39 Interestingly, HL tends to spread from the cervical nodes of one side of the neck to the mediastinum before it spreads to the contralateral cervical nodes. When laparotomy was included in the staging process (which is no longer routinely performed), the spleen was noted to be involved in 27% of patients.39 When evaluating the histologic subtypes and patterns of initial involvement, the MC and LD subtypes of HL have more widespread involvement than do the NS or LP HL subtypes (see Table 69-1).
Mediastinal disease, in addition to a predilection for certain histologic subtypes, is most common in girls older than 12 years, and in those with constitutional symptoms (also known as B symptoms).40 Mediastinal disease may appear with significant respiratory compromise due to compression of the trachea, carina, (or both), including the major bronchi.41 These patients may have dyspnea on exertion or at rest, persistent cough, or stridor. They may have recently been treated for presumed asthma or bronchiolitis, without radiographic imaging. Patients with this presentation may have a history of orthopnea and are most comfortable in an upright forward-leaning position to relieve the pressure on the airway (from the anterior mediastinal mass). The physician must be vigilant for mediastinal disease because it can be silent until a patient is sedated for a radiologic or surgical procedure. These patients may prove impossible to ventilate, even with intubation, because of distal tracheal or bronchial obstruction. It is imperative that all patients with suspected lymphoma (HL or NHL) have a chest radiograph or chest CT scan before any sedation or procedure. Signs of superior vena caval obstruction, including edema and cyanosis of the face and jugular venous distention, may also be present. Extralymphatic involvement can include the liver (the most common extralymphatic organ involved), lungs, bone, bone marrow, and skin, among other sites. Whereas bone marrow involvement is present in only 4–14% of patients overall, among those patients with stage IV disease it occurs one-third of the time.42
Most patients have no systemic symptoms at the time of initial diagnosis. About one-quarter of patients will have one or more B symptoms, defined as weight loss of more than 10% in the previous 6 months, unexplained recurrent fevers greater than 38°C, or drenching night sweats.39 Pruritus, fatigue, and anorexia are other nonspecific symptoms seen in HL patients. Laboratory findings at diagnosis are nonspecific and typically are indicative of an inflammatory process. The erythrocyte sedimentation rate (ESR), serum copper, and ferritin levels are frequently elevated and may be useful later as monitors for relapse. A high ferritin (>142 ng/mL) level or increased ESR (>50) has been associated with a worse prognosis.43,44 The lactate dehydrogenase (LDH) may be elevated as well. Although not common, leukopenia may be indicative of bone marrow involvement.42
Diagnosis
The diagnostic evaluation should include a physical examination and laboratory and radiologic studies (Box 69-1). The physical examination should be directed to the obviously involved nodal groups and also to adjacent groups, keeping in mind the natural history of HL and its propensity for contiguous spread. More than four involved nodal groups in stage II patients is associated with a worse prognosis.45 Bulky disease (nodes or nodal aggregates >10 cm and/or mediastinal tumor width more than one-third of intrathoracic width on a posteroanterior chest radiograph or CT) is associated with a worse outcome in low-stage patients, and necessitates additional therapy to achieve equivalent outcomes.46–48 Auscultation of the airway, palpation of the abdomen, and examination of distant nodal groups are all critical as well.
Staging
Further evaluation of a patient with HL is required to determine the extent of disease at diagnosis and thus the stage of disease (Table 69-2). The common staging system for HL was adopted in 1971.49 This system is based on the observation of contiguous nodal spread in HL. Patients are further divided into asymptomatic (A) and symptomatic (B) subcategories. This subclassification for symptomatic patients is based on the findings of a worse prognosis for B patients and the need for a systemic therapy approach (i.e., chemotherapy in addition to radiation). This likely reflects the finding that patients with B symptoms are more likely to have distant, widespread disease when histologically staged.50
TABLE 69-2
Ann Arbor Staging Classification for Hodgkin Lymphoma
| Stage | Definition |
| I | Involvement of a single lymph node region (I) or of a single extralymphatic organ or site (IE) |
| II | Involvement of two or more lymph node regions on the same side of the diaphragm (II) or localized involvement of an extralymphatic organ or site and its regional lymph node(s) with involvement of one or more lymph node regions on the same side of the diaphragm (IIE) |
| III | Involvement of lymph node regions on both sides of the diaphragm (III), which may be accompanied by involvement of the spleen (IIIS) or by localized involvement of an extralymphatic organ or site (IIIE) or both (IIISE) |
| IV | Disseminated (multifocal) involvement of one or more extralymphatic organs or tissues with or without associated lymph node involvement or isolated extralymphatic organ involvement with distant (nonregional) nodal involvement |
For HL, the decision for the type and intensity of therapy rests on the staging results. Traditionally, two methods of staging were used in HL patients: clinical and histologic. In the past, all patients underwent both methods. Clinical staging includes physical, laboratory, and radiologic evaluations. Histologic staging requires a staging laparotomy with splenectomy, nodal sampling, and wedge biopsies of both hepatic lobes. Radiologic evaluations continue to evolve. Lymphangiograms, once a critical component of staging in HL, have been supplanted by more modern and less invasive imaging modalities. CT examination is used most frequently.51 For those who will be treated by irradiation alone, accurate assessment of abdominal disease is critical. Staging laparotomy with splenectomy, nodal sampling, and wedge biopsies of both hepatic lobes has been shown to increase the stage of disease in up to 35% of patients initially evaluated with CT (i.e., the difference between clinical and histologic staging).52,53 This would seem to indicate that abdominal exploration is important. However, with the use of systemic chemotherapy and the de-emphasis on irradiation, this discrepancy between clinical and histologic staging no longer appears to have a significant impact on treatment or outcome.54,55
For the majority of children with HL, staging is based on clinical criteria. Laparotomy (or laparoscopy) is not encouraged or recommended. However, abdominal staging should continue to be used in patients destined to be treated with irradiation alone (although this is now rare in children) because abdominal disease would have a significant impact on planned therapy.56 Staging laparotomy (or laparoscopy) with splenectomy is not without its risks. There are the typical postoperative complications of abdominal surgery. Moreover, with splenectomy, there is a lifelong risk of overwhelming sepsis with encapsulated organisms and these patients require lifelong antibiotic prophylaxis.57 An increased risk of secondary leukemia also exists in those HL patients treated with chemotherapy who have undergone splenectomy (5.9%) compared with those who have not (0.7%) as part of their staging procedure.58–60
Nuclear medicine scans are another modality used for staging HL patients. 18flurodeoxyglocose (FDG) imaging has gradually supplanted gallium scans. Fluorodeoxyglucose-labeled positron emission tomography (FDG-PET) has been found to be more sensitive and specific than either gallium or CT.61–64 Similar to gallium scanning, it leads to a higher staging in a significant percentage of patients. FDG-PET during and after therapy has been highly predictive of patient outcome65,66 and helps to differentiate residual scar tissue from residual lymphoma,67 although false-positive findings with inflammatory conditions have been reported.66 In children, it is important also to recognize the phenomena of thymic rebound after therapy. This may result in both an enlarging mediastinal mass on CT and a positive nuclear medicine scan. An experienced radiologist will recognize this phenomenon by its timing (within the first 6 months after therapy has been completed) and by the normal (although enlarged) homogeneous appearance of the thymic tissue. However, false-negative interpretations can occur. Thus, close follow-up of these patients is critical. Finally, the bone marrow examination continues to be important, regardless of planned methods of therapy, because its involvement would upgrade the patient’s disease to stage IV status and necessitate more intensive chemotherapy.
Treatment
Principles of Radiation Therapy in the Treatment of Hodgkin Lymphoma
Despite the goal of eliminating radiation from the therapeutic regimens for children with early stage HL, it must be recognized that HL is a very radiosensitive neoplasm. A long record of efficacy exists, using radiation either alone or in combination with chemotherapy for this neoplasm. Radiation therapy has traditionally been given to the sites of disease and contiguous, clinically uninvolved, areas. This is known as extended-field irradiation. More recently, involved-field irradiation has become more widely used. This is a more attractive option when combined with chemotherapy. In children, involved-field irradiation has been shown to provide excellent local control (97%).68 A study from Germany found that not only were the remission rate and disease-free survival (DFS) no different between involved-field and extended-field irradiation, but the side effects (leukopenia, thrombocytopenia, nausea, gastrointestinal toxicity, and pharyngeal toxicity) were significantly reduced when using only involved-field irradiation.69
The use of radiation therapy alone remains an option for therapy in adults with low-stage (I to III) HL because it allows them the opportunity to avoid the toxicity associated with chemotherapy.70–73 Even if relapse occurs in those treated with radiation only, the ability to salvage a long-term cure does not appear to be compromised by delaying the use of chemotherapy until the first relapse.
Currently, however, combined-modality therapy remains the standard of care for children and adolescents with HL.74
Principles of Chemotherapy
Chemotherapy is the therapeutic backbone for children with both early and advanced-stage HL. A large number of chemotherapy combinations have been used for HL. Historically, two regimens have been the most widely and effectively used for patients with early stage HL. MOPP or ABVD was administered over a 12-month period and resulted in excellent outcomes.75,76 However, these combinations have significant long-term sequelae when administered in full doses for a year. The recognition that successful treatment with chemotherapy for children with HL would have significant impact on their quality of life and ultimate survival has led to newer combinations of chemotherapy. In general, these regimens have been variations of MOPP and ABVD. These hybrids have either replaced those agents having the worst sequelae (e.g., cyclophosphamide for nitrogen mustard) or have involved the originals being given at significantly lower doses, or both.
Newer regimens in low-stage patients have been examined with lower-dose alkylating agents, which are the causes of the majority of the long-term sequelae seen in these patients.77 In addition, the number of cycles or overall duration has been significantly decreased as well.48,78 Typically, a complete therapeutic protocol currently is given over 3 to 6 months. Radiation therapy sometimes remains a part of these regimens, although it is given at lower doses and encompasses smaller fields. In some studies, the chemotherapy regimens that have been given without irradiation have produced equivalent results to regimens with irradiation in patients with low-stage disease.79–81 For those with high-risk HL, therapeutic regimens that are response-based and intensifying in both dose and timing are showing improved outcomes over the traditional regimens, with DFS now in excess of 90%.82
Stage, Histology, and Response-Based Therapy
Until recently, therapy for HL was primarily dictated by the stage at which the child was first seen. Now histology and response to therapy are added to the equation.36,83 Those with LPHL histology and low-stage disease may be considered for further reductions in chemotherapy. If the disease is completely resected via an excisional biopsy, no further treatment may be needed. In a European study of 58 children with low-stage LPHL treated with surgery alone, outcomes were good with 67% progression free survival (PFS) for those who achieved a complete response (CR) with surgery only, and with an overall survival (OS) rate of 100%.84 A COG clinical trial is attempting to confirm the results of smaller studies showing favorable outcomes with resection alone in stage I patients with LPHL, a single involved lymph node, and a complete resection. Many regimens now incorporate this concept into their design, with fewer cycles of chemotherapy or elimination of irradiation for those with early CRs.
Currently, blood or marrow stem cell transplantation is reserved for those patients whose disease is refractory to systemic chemotherapy or who have experienced relapse. Recent trials have shown that regardless of the duration of initial remission, those who are treated with high-dose chemotherapy and stem cell rescue have less treatment failure than do those treated with conventional chemotherapy.85,86 Autologous stem cell transplant (ASCT) has become the standard of care for relapsed HL.87
Promising new therapies are being investigated, including brentuximab (SGN-35), a monoclonal antibody to CD-30 that has been linked to an antitubulin agent. In a study of 42 relapsed HL patients, 15 patients had objective responses with nine CRs.88 Other novel, targeted therapies are being studied, including rituximab, an anti-CD 20 antibody; Bortezomib, a reversible proteosome inhibitor; and histone deacetylase inhibitors.87
Results
Most patients treated with combinations of chemotherapy and radiation enter into CR (>90%).89,90 Many patients, especially those with the NS subtype, may have persistent adenopathy or mediastinal enlargement for months or years after therapy. Although most prove to be cured, close follow-up is necessary. For those who do not enter remission with today’s front-line chemotherapy/irradiation combinations, the prognosis is poor. Therapeutic intensification with subsequent stem cell transplant will likely be needed.86,91
For low-risk patients, combined-modality (chemotherapy and radiation) therapy typically results in greater than 90% five-year DFS and OS rates. For intermediate risk patients, greater than 80% 5-year EFS rates are found. For high-risk patients, significant advances have been made, with EFS and OS greater that 90% in relatively recent trials.82,92,93
Long-Term Sequelae
The concern over long-term sequelae guides much of modern therapy for HL, both in adults and particularly in children. These sequelae result from both radiation therapy and chemotherapy.94,95 The long-term sequelae of irradiation in growing children are the overriding reason for the efforts to reduce or eliminate it from therapeutic regimens. Bone irradiation may result in shortening of the clavicles in those patients receiving mantle radiation or a shortened height in those receiving radiation to the spine.96 Radiation to the neck often results in permanent hypothyroidism97 and increases the risk of thyroid cancer.98,99 If radiation is to be given to the pelvis of a female patient, consideration should be given to positioning the ovaries away from the field of irradiation.100
Second malignancies are a major concern after therapy for HL.101–103 The most frequent cause of death in long-term survivors of HL is a second malignancy.104 The relative risk of a second malignancy in HL patients has been estimated to be five- to 11-fold that of the general population.102,105 This represents a 15- to 25-year actuarial risk of 7% to 23%.102,105–107 Second malignancies are more prevalent in those with HL treated before age 21 years than in the older age groups for all tumors except lung cancer.102
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


