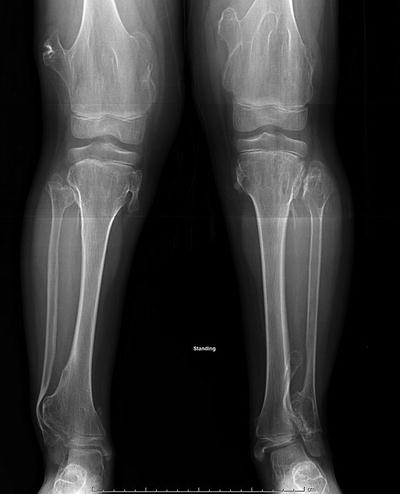
Fig. 20.1
(a) Radiograph of the ankle of a 7-year-old boy with ankle pain. Note the eccentric lobulated contained lesion which thins the surrounding cortex. Biopsy at the time of surgery confirmed the diagnosis of a nonossifying fibroma. (b) Radiograph 8 years following curettage and bone graft shows resolution of the lesion. The boy is asymptomatic and participates in contact athletics
Biopsy is often not needed since the radiographic appearance is characteristic and does not resemble malignancy. Specimens from nonossifying fibromas show sheets of bland fibroblasts with small nuclei without pleomorphism.
Treatment of nonossifying fibromas is nonsurgical in the vast majority of patients. Size greater than 50 % of the width of the bone and greater than 3.3 cm in length has been shown to be predictive of pathologic fracture, but smaller lesions have been noted to lead to stress fractures too, albeit rare [4, 5]. Pathologic fractures heal with immobilization and may lead to resolution of the lesion. If the nonossifying fibroma is in a weight-bearing bone, is sufficiently large, and has undergone pathologic fracture, curettage and bone grafting can eradicate the lesion and allow for full athletic participation [6, 7] (see Fig. 20.1b) The natural history of nonossifying fibromas is that they regress during adulthood.
Box 20.1. Nonossifying Fibroma
Age at presentation: 1st or 2nd decade.
Location: Eccentric metaphyis.
Number: solitary.
Presenting symptoms: usually none, occasional fracture.
Rx: none unless large.
Malignant degeneration: none.
Fibrous Dysplasia
Fibrous dysplasia is a noninherited condition defined by the replacement of normal trabecular bone by fibrous tissue and immature disorganized osteoid [8]. When monostotic, the condition shows no gender predilection. Fibrous dysplasia may be diagnosed in childhood, adolescence, or adulthood. Lesions may be present in one bone, known as monostotic fibrous dysplasia, or in many bones, known as polyostotic fibrous dysplasia. When associated with endocrine abnormalities such as precocious puberty, the diagnosis of McCune Albright syndrome is made. Mazabraud’s syndrome is the association of fibrous dysplasia with muscular myxomas and is even more rare. Polyostotic fibrous dysplasia is more common in girls than in boys, especially when associated with endocrinopathy.
Fibrous dysplasia and McCune Albright syndrome have been found to result from a somatic mutation in the guanine nucleotide-binding protein, Alpha-stimulating activity polypeptide gene, known more succinctly as the Gnas pathway [9, 10]. Because this is a somatic mutation, the disease is not hereditary. This mutation is specific to fibrous dysplasia and is not seen in other lytic processes of bone.
Presenting symptoms of fibrous dysplasia include limp, pathologic fracture, or bone pain [11]. Patients may present with angular deformity less frequently. Those children with McCune Albright syndrome present with precocious puberty, which usually manifests as painless vaginal bleeding in young girls. They also have characteristic café-au-lait spots on their skin, present from birth or infancy, with irregular borders described as “coast of Maine” (Fig. 20.2). Other endocrinopathies associated with McCune Albright syndrome include hyperthyroidism, growth hormone excess, and Cushing’s disease [12, 13].
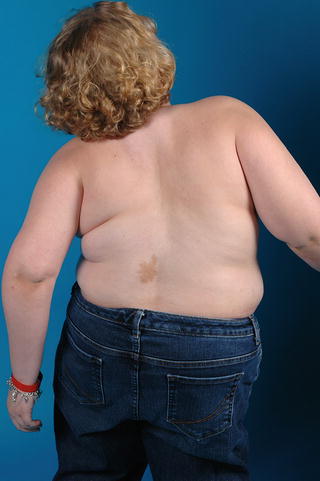
Fig. 20.2
Clinical photograph of a girl with McCune Albright syndrome. Note the skin macule which is hyperpigmented and has irregular borders. The child has spinal involvement and scoliosis
The radiographic appearance of fibrous dysplasia is one of one or more ill-defined lytic lesions. The disease may be present in the metaphysis or diaphysis of the bone, but is typically not epiphyseal. The margins of the lesions are somewhat indistinct, lacking the sclerotic rim frequently present in patients with simple bone cysts. Owing to the lack of margins, the lesions of fibrous dysplasia are described as “flame-shaped,” tapering off into the normal bone. As the bony trabeculae are replaced by fibrous tissue, the appearance within the lesions is blurred, opaque, and gray on radiographs. This has been termed “ground glass” appearance. The lesions can be expansile and may thin the cortices of the native bone. They do not create a periosteal reaction (Fig. 20.3). Large lesions in weight-bearing bones may result in bowing of the bone, with coxa vara of the femur and anterior bowing of the tibia being most common.
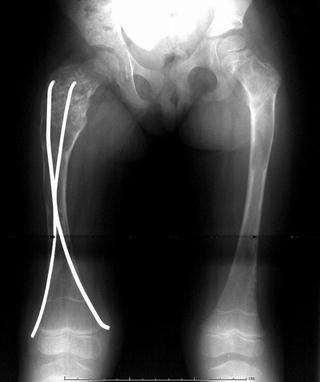
Fig. 20.3
Anteroposterior radiograph of a 3-year-old female with polyostotic fibrous dysplasia. Poorly defined lytic areas are present throughout both femora and the right hemipelvis. The right femoral neck is expanded, and an early shepherd’s crook deformity is present. Intramedullary fixation is present due to previous pathologic fracture
The most common sites for fibrous dysplasia are within the long bones, but any bone may be involved. Vertebral involvement is seen less frequently. The skull may be affected, which can lead to disfigurement over time.
The differential diagnosis in patients with monostotic fibrous dysplasia includes simple and aneurysmal bone cysts, eosinophilic granuloma, and when the tibia is involved, osteofibrous dysplasia. Lesions due to fibrous dysplasia can usually be differentiated from bone cysts as the margins of the lesion are less distinct, there is no fallen leaf sign (seen in patients with simple cysts following pathologic fractures), and the matrix radiographically is more opaque in fibrous dysplasia. Upon presentation, it has been recommended to obtain a technetium bone scan to identify the presence of polyostotic disease. By the age of 6 years, it has been found that all affected sites can be identified on bone scan [11]. Due to the lack of need for treatment of asymptomatic lesions, it has not been the practice of my institution to obtain a bone scan for identification of all bony lesions.
The diagnosis is usually made both clinically and radiographically (especially in the polyostotic form); therefore biopsy is only rarely required. When biopsy tissue is obtained, firstly, the lesion in fibrous dysplasia is filled with tissue rather than fluid on aspiration, differentiating it from bone cysts. On gross examination, tissue from fibrous dysplasia lesions appears firm and gray. There may be areas that appear cystic. The specimen will feel gritty to palpation. Microscopic examination shows the abundant fibrous tissue with limited cellularity. Islands of immature osteoid are present, but do not form mature trabeculae. These spicules of immature bone take on abnormal irregular shapes, which have been termed “Chinese Letters.” The islands of osteoid are not rimmed in osteoblasts, in distinction to osteofibrous dysplasia. Giant cells and osteoclasts may be scattered in the microscopic field.
The orthopedic management of fibrous dysplasia is aimed towards correcting deformity and the treatment of pathologic fractures. The natural history of fibrous dysplasia in the skeletally immature patient is that of slow expansion of the bony lesions. Deformity of weight-bearing bones appears as the mechanical strength of the bone is lost due to progressive replacement with fibrous tissue. In polyostotic disease, gait becomes more difficult, with the median age at which patients require assistive devices to walk being 7 years [14]. Characteristic deformities include coxa vara due to the “shepherd’s crook” deformity of the femur, angular deformity at the knee, and procurvatum of the tibiae. Pain in the extremity may occur as the bone weakens and deforms secondary to microfractures that occur due to the stress of weight bearing. Limb length discrepancy may develop.
Bony lesions do not require operative intervention unless associated with pathologic fracture or deformity. Curettage and bone grafting of fibrous dysplasia leads to recurrence of the lesions, and the bone graft (allograft or autograft) is eventually replaced again by the fibrous tissue characteristic of fibrous dysplasia. Cortical bone grafts have been used, with hopes of less resorption and greater improvement of mechanical strength of the bone [15]. Unfortunately, these grafts also resorb, although in a more delayed fashion compared to morselized bone graft.
Pathological fractures occur most frequently in the first decade of life between the ages of 6 and 10 years [16]. The femur is the most common bone to sustain a fracture. Fractures occurring through dysplastic bone may be treated nonoperatively in the upper extremity, but usually benefit from surgical treatment in the lower extremities. Load-sharing intramedullary implants allow earlier weight bearing, which is important due to the underlying bone fragility (Fig. 20.4) [17–19]. Deformity should be fully corrected, as residual angulation will predispose to future fractures.
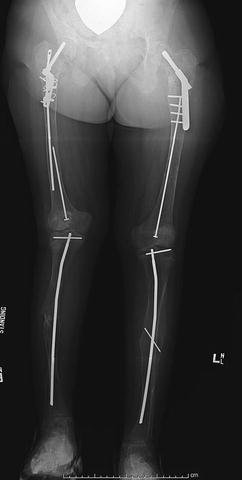
Fig. 20.4
Anteroposterior radiograph of the lower extremities of an 11-year-old girl with McCune Albright syndrome. She has undergone intramedullary fixation of the femora and tibiae as treatment of repetitive pathologic fractures. Note the proximal femoral fixation for treatment of coxa vara and shepherd’s crook deformities. There is also a leg length discrepancy
The shepherd’s crook deformity of the proximal femur is particularly challenging to treat [15, 20]. Progressive coxa vara leads to a waddling gait, hip pain, and eventual pathologic stress fractures when the neck-shaft angle is less than 120°. Proximal femoral valgus osteotomy can correct the deformity, but fixation in the dysplastic bone is extremely challenging. Plate fixation frequently leads to pathologic fracture at the ends of the plate where stress concentration occurs or to loss of fixation due to screw pullout. Hip screw and side plate fixation is typically performed; however the screw over time may migrate out of the femoral neck as the dysplastic bone remodels and the deformity recurs. Our preference is to overlap a hip screw and side plate device with an intramedullary flexible nail, with the goal of achieving reasonable proximal femoral fixation to allow the osteotomy to heal, yet providing load-sharing distal to the device by virtue of the intramedullary nail. Customized proximal femoral nail-screw devices have been used as well, but again the proximal femur typically remodels despite the presence of the implant (Fig. 20.5).
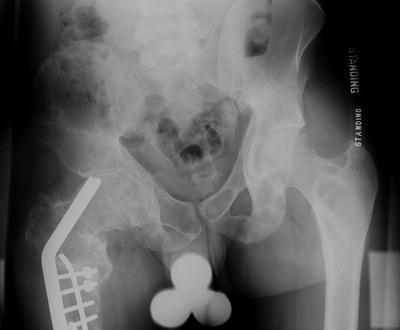
Fig. 20.5
Pelvis radiograph of a 21-year-old male who has undergone multiple surgeries for treatment of right femoral fractures and coxa vara. Note the expansion of the femoral neck, the lytic appearance of the femoral neck and head, and the superior migration of the implant in the femoral neck. There is shortening of the length of the limb on the right
Osteotomies for deformity correction require meticulous preoperative planning. The deformities are typically multiplanar, and more than one osteotomy may be required for passage of intramedullary devices. There is frequently no patent intramedullary canal in the dysplastic bone, so insertion of flexible or rigid nails must be undertaken carefully to avoid penetration with the nail or further pathologic fracture. The intramedullary canal may require drilling to allow nail passage. Additionally, large amount of blood loss should be anticipated and prepared for, as these lesions are hypervascular. Unfortunately, skeletal growth leads to recurrent deformity, and revision surgery should be expected. Progression of the fibrous dysplasia can lead to enlargement of the bone, recurrent angulation, and increasing bone pain due to bone fragility (Fig. 20.6a, b).
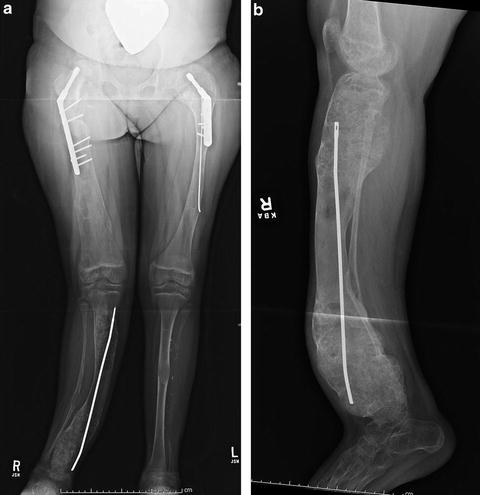
Fig. 20.6
(a) AP radiograph of the lower extremities of a 9-year-old girl with polyostotic fibrous dysplasia. She has undergone a right proximal femoral allograft replacement due to marked bony enlargement suspicious for malignancy, a left proximal femoral osteotomy, and right tibial osteotomy with intramedullary fixation. (b) Two years later, the tibia has undergone great expansion and angular deformity has worsened with recurrent bone pain with weight bearing
Nonunion is infrequent in fibrous dysplasia. Bony callous can develop, but the bone softens with time due to replacement with fibrous tissue.
Limb length discrepancy is seen most frequently in children with severe polyostotic disease. Lengthening is usually fraught with complications due to the poor mechanical properties of the dysplastic bone. Contralateral epiphysiodesis may be offered, but is often rejected by some families due to the patient’s underlying short stature.
Scoliosis is seen with greater frequency in patients with fibrous dysplasia [21, 22]. Vertebral involvement with scoliosis has been found in one-third to over 50 % of patients with polyostotic disease. As would be expected, spinal fusion surgery may be difficult due to the bony fragility and excessive bleeding. Yet successful results of posterior spinal fusion have been published [22]. The efficacy of scoliosis bracing in fibrous dysplasia has not been proven, and the concurrent presence of rib lesions precludes its use in most patients.
Medical treatment with bisphosphonate agents has been used in patients with polyostotic fibrous dysplasia [23–26]. Bisphosphonate use has not been shown to affect the bony lesions in fibrous dysplasia, but has been found to decrease the pain patients experience. New clinical studies using receptor activator of nuclear factor kappa-B ligand (RANKL) inhibitors have shown promise in influencing the progression of bone disease [27]. Patients with McCune Albright syndrome require referral at the time of diagnosis to pediatric endocrinologists for medical management [28, 29]. Those children with skull involvement should be referred as well to craniofacial specialists, as bony enlargement and overgrowth can lead to cosmetic disfigurement and optic foraminal sclerosis may progress to blindness.
The development of cystic lesions most closely resembling aneurysmal bone cysts may occur within the dysplastic bone. Rapid enlargement of the lesions coupled with increasing pain is usually present. Malignant degeneration has been documented in rare patients with fibrous dysplasia [30, 31]. Malignant degeneration is most common in adulthood and has been associated with previous irradiation, which is now contraindicated in patients with fibrous dysplasia. Painful enlargement of the dysplastic bone may also be present. In such instances, evaluation by an orthopedic oncologist should be sought.
Box 20.2. Fibrous Dysplasia
Age at presentation: 1st or 2nd decade.
Location: Long bones most frequent, metaphyis or diaphysis.
Number: solitary or polyostotic.
Presenting symptoms: limp, pathologic fracture, bone pain.
Rx: intramedullary fixation of fractures, deformity correction, bisphosphonates for pain.
Malignant degeneration: rare in adulthood.
Osteofibrous Dysplasia
Osteofibrous dysplasia, also known as Campanacci’s disease, is a peculiar bone dysplasia which is typified by both sclerotic and lytic lesions most commonly present in the tibia in children and adolescents [32, 33]. It is usually not genetically transmitted. Osteofibrous dysplasia is bimodal in presentation. It can present in early infancy with apparent anterior bowing in the tibia [34, 35]. In such cases, it must be distinguished from anterolateral bow of the tibia as associated with congenital pseudarthrosis of the tibia and neurofibromatosis. The fibula in patients with infantile osteofibrous dysplasia is usually uninvolved, and pathologic fracture is rare. The condition is nearly always unilateral and does not involve the rest of the skeleton.
The second form of osteofibrous dysplasia presents in late childhood or adolescence. It also is most common in the tibia and involves only one bone. It has been genetically linked to adamantinoma.
Osteofibrous dysplasia is not typically genetically transmitted, but a rare form has been published in which several family members were affected [36, 37].
Radiographs of osteofibrous dysplasia show bubbly lytic lesions scattered throughout the anterior diaphysis of the tibia [38]. There is usually anterior bowing, and the limb may be somewhat shorter than the contralateral leg. The cortex may appear thickened in areas (Fig. 20.7).
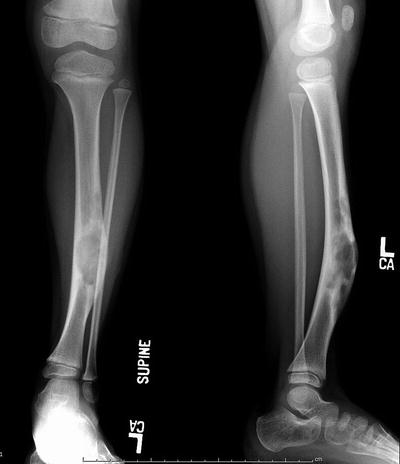
Fig. 20.7
AP and lateral radiograph of a 7-year-old female with progressive anterior bowing of the tibia. The lateral view shows loculated lytic lesions in the anterior cortex of the tibia typical for osteofibrous dysplasia
The differential diagnosis of osteofibrous dysplasia includes monostotic fibrous dysplasia, congenital pseudarthrosis of the tibia, and adamantinoma.
Pathological examination of tissue from osteofibrous dysplasia shows fibrous tissue with areas of immature osteoid, as is seen in fibrous dysplasia. The histological distinction from fibrous dysplasia is that the osteoid is rimmed in osteoblasts in osteofibrous dysplasia, which is its characteristic histologic finding. Cells stain positively for cytokeratin, which is also present in adamantinoma, supporting a relationship between the two lesions [38–42].
Treatment of osteofibrous dysplasia is controversial. Spontaneous resolution of osteofibrous dysplasia in young babies has been described, so treatment in these infants should be observation in the absence of pathologic fracture [43]. If fracture does occur, intramedullary fixation such as is performed in congenital pseudarthrosis of the tibia, combined with bone grafting the fracture site, usually is successful in obtaining and maintaining union [44].
Older children with osteofibrous dysplasia have undergone various forms of treatment. Observation of radiographically stable lesions may be chosen when the patient is asymptomatic. Marginal resection with curettage has been shown to lead to recurrence and is not recommended. Those surgeons who have concern that osteofibrous dysplasia may evolve into malignant adamantinoma have performed aggressive resection of the affected tibial area, with reconstruction as needed. Bone segment transport with proximal corticotomy and external fixation and treatment with free vascularized fibular graft have met with success in very rare instances [45, 46].
Box 20.3. Osteofibrous Dysplasia
Age at presentation: infantile or 1st or 2nd decade.
Location: anterior tibial diaphysis.
Number: solitary.
Presenting symptoms: bowing, enlargement, pain.
Rx: controversial, possible en bloc excision.
Malignant degeneration: associated with adamantinoma.
Enchondromatosis
Enchondromas are benign hyaline cartilaginous lesions present within the metaphysis. Enchondromas may be solitary or multiple, in which case the condition is named Ollier’s disease. Solitary enchondromas are common and rarely symptomatic, where Ollier’s disease is rare. Maffucci’s syndrome is defined as the presence of multiple enchondromas with soft tissue hemangiomatosis and carries a much bleaker prognosis. Enchondromatosis is not genetically transmitted but may be the result of a somatic mosaic mutation in isocitrate dehydrogenase 1 or 2 [47]. The incidence of Ollier’s disease is estimated at 1 per 100,000.
Presenting complaints in multiple enchondromatosis are usually bone pain due to mechanical insufficiency, pathologic fracture through the lesions, angular deformity at the knee or fingers, or limb length discrepancy [48]. The condition typically presents in the first or second decade of life.
The radiographic appearance of multiple enchondromatosis is quite distinct (Fig. 20.8). Streaky lytic lesions are seen in the metaphysis of the bones. At the knee, the elongated areas of enchondroma give a “fanlike” appearance to the metaphysis. There are areas of fine calcification within the lesions due to the presence of calcification in the cartilaginous lesions. The lesions seem to stop at the physis. It is common that multiple enchondromas are seen, and that they tend to present unilaterally. The cortex of the bone is intact in the absence of pathologic fracture. Angular deformity at the knee and limb length discrepancy can be appreciated (Fig. 20.9) [49].
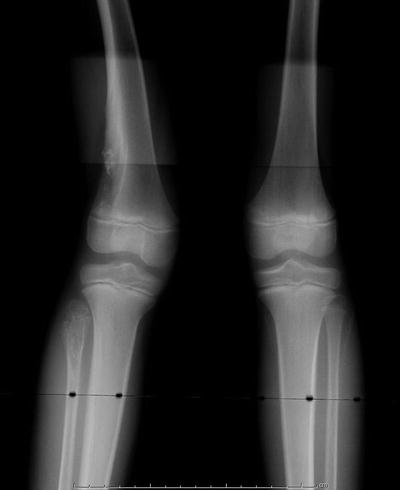
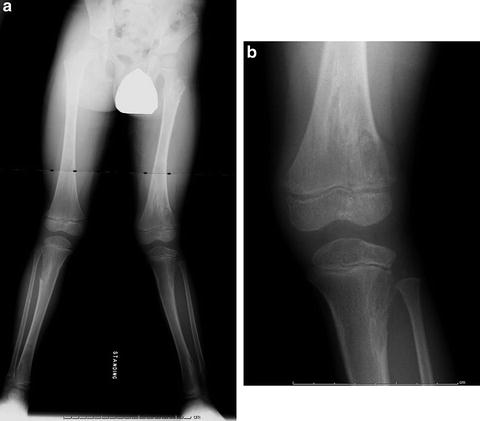
Fig. 20.8
Radiographic appearance of enchondromatosis of the distal femur and proximal tibia in a 7-year-old male. Note the eccentric location of the metaphyseal femoral lesion, and the presence of intralesional calcification which is typical of cartilage tumors. The right proximal fibula is also involved
Fig. 20.9
(a) Asymmetric genu valgum and limb length discrepancy in a young boy with Ollier’s disease. (b) A close-up of the left knee shows the streaky lytic lesions characteristic of enchondromatosis
Biopsy reveals a pearly white to bluish-white specimen. Microscopic examination shows bland cartilaginous tissue, with nests of plump chondrocytes. The nuclei are uniform, and mitotic figures may be seen in children but should be few in number. “Chicken-wire” calcification, which are areas of punctate calcification within the lesion, can be appreciated, which renders the specimen slightly gritty.
Treatment of children with solitary enchondromas is merited if there is angular deformity. Small asymptomatic lesions in the lower extremities may be observed. Multiple enchondromatosis presents for orthopedic treatment more frequently. Curettage and bone grafting of the enchondromas is in large part ineffective due to the extensive nature of the disease, but may be of some benefit in the hand. Children with Ollier’s disease have significant angular deformities, with a predisposition towards distal femoral varus [49]. The angular deformity is usually accompanied by varying amounts of limb shortening which may measure up to 25 cm at maturity (Fig. 20.10) [48]. In the lower extremity, angular correction with or without lengthening may be undertaken. Standard fixation with internal devices such as plates and screws may be compromised by poor bony integrity due to the large cartilaginous lesions. Gradual deformity correction using external fixation devices with fine wire bony fixation has been successful [50–54]. When pins or wires are inserted intralesionally, fixation can usually be maintained; however it should be expected that the regenerate will contain enchondromatosis tissue (Fig. 20.11a, b) [53]. Lengthening with intramedullary devices has also been reported [55].
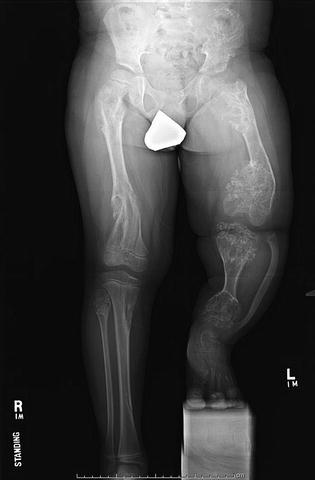
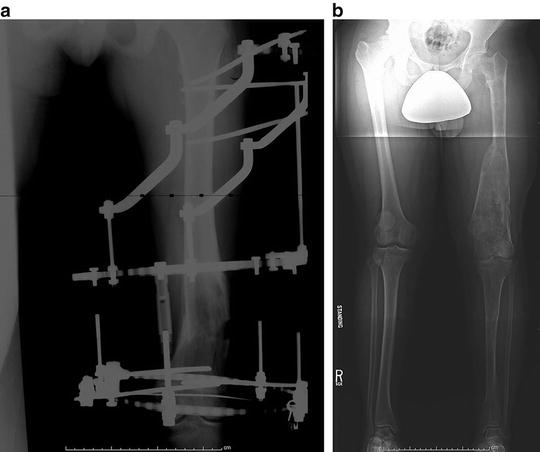
Fig. 20.10
Severe limb length discrepancy measuring greater than 15 cm and varus deformity of the left distal femur in 7-year-old male with Ollier’s disease. Note the presence of enchondromas in bilateral femora and tibias as well as the pelvis. The extent of involvement is asymmetric
Fig. 20.11
(a) Varus osteotomy of the distal femur in patient with Ollier’s disease. External fixation with half-pins and fine wires was successful in stabilizing the osteotomy. (b) Standing radiograph 1.5 years following removal of the external fixator shows satisfactory alignment but a persistent limb length discrepancy
Malignant transformation has been described in enchondromatosis in up to 20–30 % of patients by the age of 40. Patients with significant expansion of a lesion or with increasing pain should be suspected. Chondrosarcoma is the most frequent malignant tumor seen in up to 25 to 30 % of adult patients with enchondromatosis [56, 57]. Histologic overlap between benign enchondromas seen in Ollier’s disease and low-grade chondrosarcoma makes the diagnosis of malignant degeneration challenging. Remote non-bony malignancies are associated with Maffucci’s syndrome, which carries a nearly 100 % incidence of the development of a malignant tumor [58]. These children must be carefully monitored throughout their lifetime for the appearance of cancerous lesions.
Box 20.4. Enchondroma
Age at presentation: 1st or 2nd decade.
Location: metaphyis, predisposition to asymmetry.
Number: solitary or multiple (Ollier’s disease).
Presenting symptoms: bone pain, pathologic fracture, angular deformity, limb length discrepancy.
Rx: osteotomy for deformity correction, lengthening, in hand may curettage and bone graft.
Malignant degeneration: chondrosarcoma in 25–30 % of Ollier’s disease.
Solitary Osteochondromas and Multiple Hereditary Exostoses
An osteochondroma is a common benign bone growth which protrudes from the surface of the native bone and is capped with cartilage. Osteochondromas occur in the metaphyseal area and have been hypothesized to be the result of sequestration of a fragment of physis which causes the outgrowth. The bone of an osteochondroma is normal bone and is in continuity with the host bone. Osteochondromas may occur in any bone, including the pelvis, spine, and fingers, but are most common in the distal femur, proximal tibia, and proximal humerus [59].
Multiple hereditary osteochondromatosis or exostoses (MHE) are an inherited condition typified by the presence of multiple osteochondromas scattered throughout the skeleton. It is genetically transmitted as an autosomal dominant trait with very high (96 %) penetrance but variable phenotypic expression. The number and location of osteochondromas can vary among family members. The tumor suppressor genes EXT1, EXT2, and EXT3 for MHE have been localized on chromosomes 8, 11, and 19 [60–62]. Patients with MHE have a loss of function of these genes, thereby allowing growth of the osteochondromas. Patients with EXT1 mutations generally have more severe phenotypical expression and shorter stature than those with EXT2 mutations [63–67]. It is believed that patients with solitary osteochondromas have somatic, but not generalized, mutations in the EXT gene. The incidence of multiple osteochondromas is 1 per 50,000. Ninety percent of patients with MHE have a positive family history and demonstrable mutations in the EXT 1 or 2 genes [68].
Patients with a single osteochondroma often present in the first or second decade for evaluation of a painless periarticular mass. Patients with MHE usually present in early childhood with multiple small masses. The median age of presentation in MHE is 3 years [69]. The presence of a positive family history generally created a heightened awareness of the condition in the parents and therefore an earlier presentation. Patients may complain of pain if there has been trauma in the area of the osteochondroma, or if the mass is sufficiently large to create symptoms due to impingement of overlying muscle or tendons. In many cases, the patient is asymptomatic, but the lesion has been identified coincidentally on a radiograph obtained for another reason. Patients with MHE tend to be of mildly short stature. Stature less than the 25th percentile was seen in 58 % of patients with MHE [66]. Additionally, the extremities will appear shorter than the trunk does, with the amount of shortening being linked to the size of osteochondromas present in the limb segment [70]. Angular deformity, such as genu valgum and ankle valgus, may be present.
Radiographs show a bony protrusion in the periarticular area originating from the metaphyseal bone (Fig. 20.12). Osteochondromas are not epiphyseal. The cortex of the osteochondroma is in continuity with the native bone, and the trabeculae within the osteochondroma are similarly in continuity with the metaphysis. Osteochondromas may be pedunculated, with a distinct bony stalk, or sessile, where the lesion is broad-based. When pedunculated, osteochondromas typically point away from the joint. Osteochondromas may vary in size. Large osteochondromas in two-bone segments, such as the lower leg, can cause deformity of the smaller bone, in this case the fibula. Osteochondromas in the distal femur, proximal tibia, and/or proximal fibula are seen in 94 % of children with MHE. Proximal humerus masses are present in 50 % of cases, the scapula and ribs in 40 %, the distal radius and ulna in 30 %, the proximal femur in 30 %, the phalanges in 30 %, the distal fibula in 25 %, and the distal tibia in 20 % of MHE patients [69].