Chapter 13
Local Anesthetics and Opioids
Brenda A. Bucklin MD, Alan C. Santos MD, MPH
Chapter Outline
Local anesthetics and opioids are often used for pain relief in obstetric practice. Local anesthetics may be used for infiltration anesthesia, peripheral (pudendal) nerve block, or neuraxial block, whereas opioids are administered both systemically and neuraxially. The physiologic changes that occur during pregnancy may affect the pharmacology of both local anesthetics and opioids. In turn, these analgesic drugs may have effects on the mother and the fetus.
Local Anesthetics
Molecular Structure
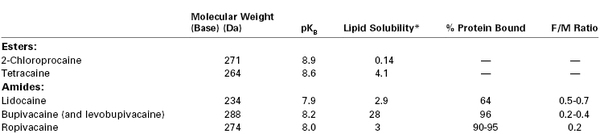
All local anesthetic molecules except cocaine contain a desaturated carbon ring (aromatic portion) and a tertiary amine connected by an alkyl chain (Figure 13-1). The intermediate alkyl chain, by virtue of its ester or amide linkage, is the basis for the classification of local anesthetics as amino-esters (which are hydrolyzed by pseudocholinesterase) and amino-amides (which undergo hepatic microsomal metabolism) (Table 13-1). The aromatic ring of the esters, which renders the molecule lipid soluble, is a derivative of benzoic acid. The amide aromatic ring is a homologue of aniline. The tertiary-amine portion acts as a proton acceptor; thus, local anesthetics behave as weak bases. In its quaternary (i.e., “protonated”) form, the terminal amine is the water-soluble portion. The Henderson-Hasselbalch equation predicts the relative proportions of local anesthetic that exist in the ionized and un-ionized form. The higher the pKB (base dissociation constant) relative to physiologic pH, the smaller the proportion of drug that exists in the un-ionized form. All amide local anesthetics (with the exception of lidocaine) exist as stereoisomers because of the presence of an asymmetric carbon adjacent to the terminal amine.
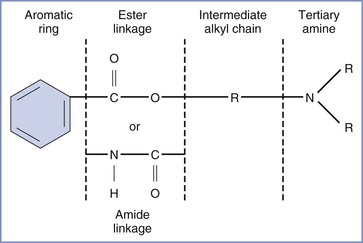
FIGURE 13-1 Structure of the molecule of a local anesthetic. R, alkyl group. (Modified from Santos AC, Pedersen H. Local anesthetics in obstetrics. In Petrie RH, editor. Perinatal Pharmacology. Cambridge, MA, Blackwell Scientific, 1989:373.)
Clinical formulations of local anesthetics are prepared as hydrochloride salts to increase their solubility in water. These solutions are usually acidic (i.e., pH of 4 to 6) to enhance formation of the water-soluble quaternary amine and to prevent oxidation of the epinephrine present in epinephrine-containing solutions.
Chirality
With the exception of lidocaine, amide local anesthetics are known as chiral compounds because they have a single asymmetric carbon adjacent to the amino group and thus exist in isomeric forms that are mirror images of each other. The direction in which the isomers rotate polarized light distinguishes them as either dextrorotary (D) or levorotary (L) isomers. This distinction is important, because individual isomers of the same drug may have different biologic effects. As a rule, the levorotary isomer of a drug has greater vasoconstrictor activity and a longer duration of action but less potential for systemic toxicity than the dextrorotary form.1
In the past, single-isomer formulations were costly to produce; and for that reason, local anesthetics used clinically (e.g., bupivacaine) have contained a racemic mixture of both the dextrorotary and levorotary forms of the drug. However, with improved techniques of selective extraction, two commercially available single-isomer formulations of local anesthetic are now available, ropivacaine and levobupivacaine. Levobupivacaine is the levorotary isomer of bupivacaine; it is currently not marketed in the United States. Ropivacaine is a homologue of mepivacaine and bupivacaine, but, unlike these other local anesthetics, it is formulated as a single levorotary isomer rather than as a racemic mixture. A propyl group on the pipechol ring distinguishes ropivacaine from bupivacaine (which has a butyl group) and mepivacaine (which has a methyl group).2 Thus, it is not surprising that the physicochemical characteristics of ropivacaine are intermediate between those of mepivacaine and bupivacaine.
The reduction in systemic toxicity observed with administration of the levorotary isomers may be both drug and concentration dependent. For example, one study in isolated guinea pig hearts noted that bupivacaine isomers lengthened atrioventricular conduction time more than ropivacaine isomers did. In contrast to other measured variables, “atrioventricular conduction time showed evident stereoselectivity” for bupivacaine at the lowest concentration studied (0.5 µM) but only at much higher concentrations for ropivacaine (> 30 µM).3
Mechanism of Action
At rest, the interior of a nerve cell is negatively charged in relation to its exterior. This resting potential of 60 to 90 mV exists because the concentration of sodium in the extracellular space greatly exceeds that in the intracellular space. The converse is true for potassium. Excitation results in the opening of membrane channels, which allows sodium ions to flow freely down their concentration gradient into the cell interior. Thus, the electrical potential within the nerve cell becomes less negative until, at the critical threshold, rapid depolarization occurs. This depolarization is needed to initiate the same sequence of events in adjacent membrane segments and for propagation of the action potential. Thereafter, sodium channels close and the membrane once again becomes impermeable to the influx of sodium. The negative resting membrane potential is reestablished as sodium is removed from the cell by active transport. At the same time, potassium passively accumulates within the resting cell.
Interference with sodium-ion conductance appears to be the mechanism by which local anesthetics reversibly inhibit the propagation of the action potential. Four major theories attempt to explain this effect. The most prominent hypothesis is that the local anesthetic interacts with receptors in the nerve cell membrane that control channels involved in sodium conductance.4 There may be more than one site at which local anesthetics bind to sodium-channel receptors (Figure 13-2).
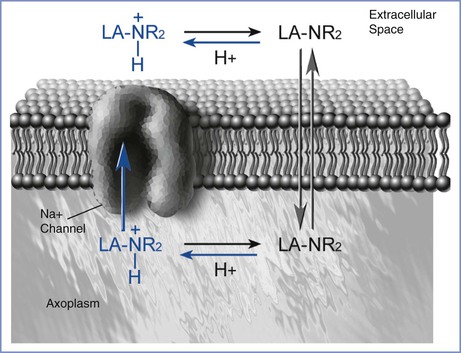
FIGURE 13-2 Local anesthetic access to the sodium channel. The uncharged molecule (LA-NR2) diffuses most easily across the lipid membrane and interacts with the sodium channel at an intramembranous site. The charged molecule (LA-NR2H+) gains access to a specific receptor on the sodium channel in the intracellular space. (Drawing by Naveen Nathan, MD, Northwestern University Feinberg School of Medicine, Chicago, IL.)
The Meyer-Overton theory offers a second explanation for local anesthetic action. This hypothesis suggests that the lipid-soluble portion of the local anesthetic molecule expands the cell membrane and interferes with rapid sodium conductance. A third possibility is that local anesthetics may alter the membrane surface charge, a change that would inhibit propagation of the action potential. Fourth, local anesthetics may displace calcium from sites that control sodium conductance.
Both the un-ionized and ionized forms of a local anesthetic are involved in pharmacologic activity. The un-ionized base, which is lipid soluble, diffuses through the cell membrane, whereas the charged form is much more active in blocking the sodium channel.
Pharmacodynamics
Pregnant women typically require smaller doses of local anesthetic compared with nonpregnant women for neuraxial blockade. This effect may be evident as early as the second trimester.5,6 This difference has been attributed to enhanced spread of local anesthetic due to epidural venous engorgement. However, mechanical effects alone do not account for the observation that the spread of spinal and epidural analgesia in early pregnancy is similar to that in pregnant women at term.5–7 In fact, pregnancy may also enhance neuronal sensitivity to local anesthetics. For example, pregnancy increases median nerve susceptibility to lidocaine.8 In vitro studies demonstrated that the onset of neural blockade was faster, and lower concentrations of bupivacaine were required to block vagal fibers, in pregnant rabbits than in nonpregnant rabbits.9
Hormonal and biochemical changes may be responsible for the greater susceptibility to neural blockade during pregnancy. For example, one study demonstrated an enhanced effect of bupivacaine in isolated vagus fibers from nonpregnant, ovariectomized rabbits who had received long-term (4 days) but not short-term exposure to progesterone.10 A higher pH and lower bicarbonate and total carbon dioxide content have been demonstrated in cerebrospinal fluid (CSF) from women undergoing cesarean delivery than in CSF from age-matched nonpregnant controls. A higher pH increases the proportion of local anesthetic that exists as the base form and facilitates diffusion of the drug across nerve membranes.7
Pharmacokinetics
Pregnancy is associated with progressive physiologic adaptations that may influence drug disposition (see Chapter 2). However, it is difficult to predict with certainty the effects of pregnancy on the pharmacokinetics of an individual drug.
2-Chloroprocaine
2-Chloroprocaine is hydrolyzed rapidly by plasma pseudocholinesterase to chloroaminobenzoic acid and H2O. The in vitro half-life of 2-chloroprocaine in sera from men is less than 15 seconds.11 Although pregnancy is associated with a 30% to 40% decrease in pseudocholinesterase activity, the half-life of 2-chloroprocaine in maternal plasma in vitro is 11 to 21 seconds. After epidural injection, the half-life of 2-chloroprocaine in the mother ranges from 1.5 to 6.4 minutes.12 The longer half-life after epidural administration results from continued absorption of the drug from the injection site. Administration of 2-chloroprocaine to patients with low pseudocholinesterase activity may result in prolonged local anesthetic effect and a greater potential for systemic toxicity.13
Lidocaine
The volume of the central compartment and the volume of distribution are greater in pregnant ewes than in nonpregnant ewes.14,15 Bloedow et al.15 observed that the total body clearance of lidocaine was similar in the two groups of animals. They concluded that the elimination half-life of lidocaine, which depends on the balance between volume of distribution and clearance, was longer in pregnant ewes.15 In contrast, Santos et al.14 concluded that the elimination half-life of lidocaine was similar in the two groups of sheep because the total body clearance of the drug was greater in pregnant animals than in nonpregnant animals. This discrepancy could result from differences in the complexity of the surgical preparation and the allowed recovery period. In pregnant women, the elimination half-life of lidocaine after epidural injection is approximately 114 minutes.16
Lidocaine is metabolized to two active compounds, monoethylglycinexylidide (MEGX) and glycinexylidide (GX). Monoethylglycinexylidide can be detected in maternal plasma within 10 to 20 minutes after neuraxial injection of lidocaine, whereas glycinexylidide can be detected within 1 hour of epidural injection but rarely after subarachnoid injection.17,18 Urinary excretion of unchanged lidocaine is negligible in sheep (i.e., < 2% of the administered dose) and is not affected by pregnancy.14
The physiologic changes that occur during pregnancy are progressive. However, little information is available about the pharmacokinetics of local anesthetics before term. In one study, total clearance of lidocaine was similar at 119 and 138 days’ gestation in gravid ewes (term is 148 days).19
Lidocaine is predominantly bound to alpha1-acid glycoprotein (AAG) in plasma.20 Pregnancy leads to a decreased concentration of AAG; thus, the free plasma fraction of lidocaine is higher in term pregnant women than in nonpregnant controls.20 The increase in the free fraction of lidocaine occurs early in gestation and is progressive.21
Bupivacaine
At least two studies compared the pharmacokinetics of bupivacaine after epidural administration in pregnant and nonpregnant women.22,23 The absorption rate, the area under the concentration-time curve, and the elimination half-life (12 to 13 hours) were similar in the two groups. The elimination half-life of bupivacaine after epidural administration is much longer than that reported after intravenous injection, largely because the drug is continuously absorbed over time from the epidural space.
After intravenous injection, the volume of distribution of bupivacaine is lower in pregnant sheep than in nonpregnant sheep.24 In contrast, ovine pregnancy is associated with a greater volume of distribution of lidocaine.14,15 The differences in gestational effects on the volume of distribution of the two local anesthetics may result from the greater binding of bupivacaine to plasma proteins during gestation (whereas the converse occurs with lidocaine).24 In one study, urinary excretion of unchanged bupivacaine was not affected by pregnancy and was less than 1% of the administered dose.22 Nonetheless, low concentrations of bupivacaine may be detected in the urine of pregnant women for as long as 3 days after delivery.25
Bupivacaine undergoes dealkylation in the liver to 2,6-pipecolyxylidide (PPX). After epidural injection of bupivacaine for cesarean delivery, PPX was detected in maternal plasma within 5 minutes and remained detectable for as long as 24 hours.25 With the lower doses required for labor analgesia, PPX was found only if the block was maintained with multiple reinjections during a period that exceeded 4 hours.26 Pregnancy may affect metabolism of bupivacaine.22 For example, pregnant women have higher serum PPX concentrations, but the unconjugated 4-hydroxy metabolite is not produced in significant amounts. The reason for this finding is unclear but may be related to the effects of hormonal changes on hepatic enzyme systems. Both progesterone and estradiol are competitive inhibitors of microsomal oxidases, whereas reductive enzymes are induced by progesterone.24 Bupivacaine is bound extensively to AAG and albumin.27 This protein binding is reduced during late pregnancy in humans.28
Long-acting pipechol amide local anesthetics, such as bupivacaine, are beneficial for neuraxial labor analgesia because they produce a relative motor-sparing block as compared with other local anesthetics. The effective dose in 50% of cases (ED50) for motor block after intrathecally administered bupivacaine was lower in pregnant than in nonpregnant women (3.96 mg and 4.14 mg, respectively).29
Ropivacaine
Pregnant sheep have a smaller volume of distribution and a slower clearance of ropivacaine than nonpregnant animals.24 However, the relationship between volume of distribution and clearance is such that the elimination half-life is similar in pregnant and nonpregnant animals.
After intravenous injection in laboratory animals, the elimination half-life of ropivacaine is shorter than that of bupivacaine.24,30 Similar findings have been described after intravenous injection in nonpregnant human volunteers.31 The shorter elimination half-life of ropivacaine has been attributed to a faster clearance and a shorter mean residence time than for bupivacaine.24
Peak plasma concentration (Cmax) after epidural administration of 0.5% ropivacaine and 0.5% bupivacaine for cesarean delivery are similar (1.3 µg/mL and 1.1 µg/mL, respectively).32 The elimination half-life of ropivacaine is 5.2 ± 0.6 hours, which is shorter than that for bupivacaine, at 10.9 ± 1.1 hours. No difference in clearance between the two drugs has been noted.
Like bupivacaine, ropivacaine is metabolized by hepatic microsomal cytochrome P450. The major metabolite is PPX, and minor metabolites are 3′- and 4′-hydroxy-ropivacaine.33
Ropivacaine is highly bound (approximately 92%) to plasma proteins but less so than bupivacaine (96%).34 Indeed, at plasma concentrations occurring during epidural anesthesia for cesarean delivery, the free fraction of ropivacaine is almost twice that of bupivacaine.32 In sheep, pregnancy is associated with a greater binding of ropivacaine (and bupivacaine) to plasma proteins.24 In pregnant women undergoing epidural analgesia, the free fraction of ropivacaine decreases as the concentration of AAG increases, up to the point at which the receptors are saturated.35 However, there is little correlation between the free fraction and umbilical cord blood levels of ropivacaine at delivery.35
Effect of Histamine (H2)-Receptor Antagonists
Histamine (H2)-receptor antagonists (e.g., cimetidine, ranitidine, famotidine) are administered to increase gastric pH and reduce the parturient’s risk for aspiration pneumonitis. Drug disposition may be affected by binding to hepatic cytochrome P450, thereby reducing hepatic blood flow and renal clearance, especially with cimetidine. However, short-term administration of H2-receptor antagonists does not alter the pharmacokinetics of amide local anesthetics in pregnant women.36,37
Effects of Preeclampsia
Pathophysiologic changes associated with preeclampsia (e.g., reduced hepatic blood flow, abnormal liver function, decreased intravascular volume) may also affect maternal blood concentrations of local anesthetics (see Chapter 36). For example, Ramanathan et al.38 found that total body clearance of lidocaine after epidural injection was significantly lower in preeclamptic women than in normotensive women; however, the elimination half-life of lidocaine was similar in the two groups. Nonetheless, decreased clearance may result in greater drug accumulation with repeated injections of lidocaine in women with preeclampsia. In contrast, long-acting amides have a relatively low hepatic extraction, and changes in liver blood flow with preeclampsia may have less effect on the metabolic clearance.
Effect of Diurnal Variation
Pain may exhibit temporal variation in intensity due to diurnal neuroendocrine or external factors. Further, the pharmacokinetics and pharmacodynamics of local anesthetics may exhibit temporal patterns (i.e., chronobiology). In one study, the duration of action of epidural bupivacaine was approximately 25% longer when it was administered between 7:00 AM and 7:00 PM than between 7:00 PM and 7:00 AM.39 In contrast, another study found no diurnal variation with intrathecal bupivacaine administered for labor analgesia.40 The authors suggested that observed temporal differences in duration of analgesia may be explained by external influences such as shift changes for nurses and anesthesiologists.40
Toxicity
Systemic absorption or intravascular injection of a local anesthetic may result in local anesthetic systemic toxicity (LAST). Toxicity most often involves the central nervous system (CNS), but cardiovascular toxicity also may occur. Less common are tissue toxicity and hypersensitivity reactions.
Central Nervous System Toxicity
The severity of CNS effects is proportional to the blood concentration of local anesthetic. This relationship is well described for lidocaine (Figure 13-3). Initially, the patient may complain of numbness of the tongue, tinnitus, or lightheadedness. At high plasma concentrations, convulsions occur because of a selective blockade of central inhibitory neurons that leads to increased CNS excitation.41 At still higher concentrations, generalized CNS depression or coma may result from reversible blockade of both inhibitory and excitatory neuronal pathways. Finally, depression of the brainstem and cardiorespiratory centers may occur.
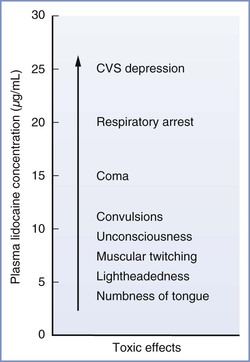
FIGURE 13-3 Signs and symptoms of systemic toxicity with increasing lidocaine concentrations. CVS, cardiovascular system. (Modified from Carpenter RL, Mackey DC. Local anesthetics. In Barash PG, Cullen BF, Stoelting RK, editors. Clinical Anesthesia. Philadelphia, Lippincott, 1992:527.)
The relative toxicity of a local anesthetic correlates with its potency. For lidocaine, etidocaine, and bupivacaine, the ratio of the mean cumulative doses that cause convulsions in dogs is approximately 4 : 2 : 1, which is similar to their relative anesthetic potencies.42 The same relative toxicity was demonstrated in human volunteers.43 Local anesthetics may be ranked in order of decreasing CNS toxicity as follows: bupivacaine, ropivacaine, levobupivacaine, lidocaine, and 2-chloroprocaine.44 Tetracaine, etidocaine, and mepivacaine are used rarely in obstetric anesthesia practice.
Other factors (e.g., the speed of injection) may affect CNS toxicity. In humans, the mean dose of etidocaine that elicited signs of CNS toxicity was lower during a 20-mg/min infusion than during a 10-mg/min infusion.43 The seizure threshold also may be affected by metabolic factors. For example, in cats, an increase in PaCO2 or a decrease in pH results in a reduction in the seizure-dose threshold for local anesthetics. Respiratory acidosis may result in delivery of more drug to the brain; alternatively, respiratory acidosis may result in “ion trapping” of the local anesthetic and/or an increase in the unbound fraction of drug available for pharmacologic effect.45–47
Cardiovascular Toxicity
The cardiovascular system is much more resistant than the CNS to the toxic effects of local anesthetics. Severe, direct cardiovascular depression is rare, especially in association with the use of lidocaine. Prompt administration of oxygen and, if necessary, initiation of ventilatory and circulatory support usually prevent cardiac arrest after unintentional intravenous injection of lidocaine.48 Progressive depression of myocardial function and profound vasodilation occur only at extremely high plasma concentrations.48 In contrast, the more potent amide local anesthetics (i.e., bupivacaine) have a more narrow margin of safety, expressed as the ratio between the dose (or plasma concentration) required to produce cardiovascular collapse and the dose (or plasma concentration) required to produce convulsions.48 A partial explanation is the fact that supraconvulsant doses of bupivacaine (but not of lidocaine) precipitate lethal ventricular arrhythmias.49–51 These arrhythmias may be caused by exaggerated electrophysiologic effects (e.g., depression of ventricular conduction) out of proportion to bupivacaine’s anesthetic potency.52
Two theories have been proposed to explain why malignant ventricular arrhythmias occur with bupivacaine but not with lidocaine. Both bupivacaine and lidocaine rapidly block cardiac sodium channels during systole, but bupivacaine dissociates from these channels during diastole at a much slower rate than lidocaine.52 Thus, at physiologic heart rates, the diastolic period is of sufficient duration for lidocaine to dissociate from sodium channels, whereas a bupivacaine block becomes intensified. This difference makes bupivacaine much more potent than lidocaine in depressing conduction and inducing reentrant-type ventricular arrhythmias. Alternatively, other investigators have suggested that high concentrations of local anesthetic in the brainstem may lead to systemic hypotension, bradycardia, and ventricular arrhythmias.53 These effects occur more commonly with bupivacaine because of its high lipid solubility, which facilitates transfer across the blood-brain barrier. An echocardiographic study in anesthetized dogs suggested that bolus injection of bupivacaine results in systolic dysfunction, especially involving the right ventricle, which precedes the occurrence of arrhythmias.54
Systemic Toxicity of Ropivacaine and Levobupivacaine
In perfused preparations of myocardium, ropivacaine is intermediate between bupivacaine and lidocaine in its depressant effect on cardiac excitation and conduction as well as in its potential to induce reentrant-type ventricular arrhythmias.34 In dogs, the margin of safety between convulsive or lethal doses and plasma concentrations of drug is greater for ropivacaine than for bupivacaine but less than that for lidocaine.55 The arrhythmogenicity of ropivacaine in pigs also is intermediate between that of lidocaine and bupivacaine.56 In sheep, the ratio of fatal doses of bupivacaine, ropivacaine, and lidocaine is 1 : 2 : 9.57 Ropivacaine was found to cause fewer CNS symptoms and was 25% less toxic than bupivacaine (as defined by the doses and plasma concentrations that were tolerated) when administered to healthy male volunteers.58
Most studies comparing the systemic toxicity of ropivacaine and bupivacaine have used equal doses of each, and, therefore, cannot resolve the controversy as to whether ropivacaine truly is less cardiotoxic or merely less potent than bupivacaine. This issue would be of concern only if larger doses of ropivacaine than bupivacaine were required to produce comparable regional blocks. Indeed, several studies in laboring women suggest that ropivacaine is 25% to 40% less potent than bupivacaine.59–61 Thus, the need for a larger dose of ropivacaine may negate the expected benefits of its apparently wider margin of safety. However, results from one laboratory study showed that ropivacaine produces less cardiotoxicity than bupivacaine, even when given at equipotent doses.62
Long-acting amide local anesthetics—even the newer drugs—are very potent and may cause cardiac arrest with a misplaced injection or relative overdose. Indeed, several cardiac arrests have been reported with the use of ropivacaine,63,64 including one in a woman undergoing a cesarean delivery with epidural anesthesia.65 In contrast to that induced by bupivacaine,66 resuscitation from a cardiac arrest induced by ropivacaine may be successful more often than not.63–65
Evidence suggests that levobupivacaine causes fewer arrhythmias than the racemic drug. Valenzuela et al.67 demonstrated that levobupivacaine caused less inhibition of inactivated sodium channels than either the dextrorotary or racemic drug. In comparison with dextrorotary and racemic bupivacaine, levobupivacaine resulted in less QRS widening and a lower frequency of malignant ventricular arrhythmias in isolated, perfused rabbit hearts.68 Similarly, levobupivacaine produced less second-degree heart block and atrioventricular conduction delay than the other two forms of the drug in isolated perfused guinea pig hearts.3
In laboratory animals, the systemic toxicity of levobupivacaine is intermediate between that of bupivacaine and ropivacaine.69
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


