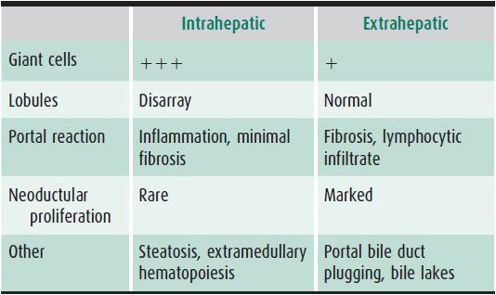
Table 22–2. Characteristic histologic features of intrahepatic and extrahepatic neonatal cholestasis.
INTRAHEPATIC CHOLESTASIS
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Elevated total and conjugated bilirubin.
Elevated total and conjugated bilirubin.
 Hepatomegaly and dark urine.
Hepatomegaly and dark urine.
 Patency of extrahepatic biliary tree.
Patency of extrahepatic biliary tree.
 General Considerations
General Considerations
Intrahepatic cholestasis is characterized by impaired hepatocyte secretion of bile and patency of the extrahepatic biliary system. A specific cause can be identified in about 60% of cases. Patency of the extrahepatic biliary tract is suggested by pigmented stools and lack of bile duct proliferation and portal tract bile plugs on liver biopsy. It can be confirmed least invasively by hepatobiliary scintigraphy using technetium-99m (99mTc)-dimethyliminodiacetic acid (diethyl-IDA [DIDA]). Radioactivity in the bowel within 4–24 hours is evidence of bile duct patency, as is finding bilirubin in duodenal aspirates. However, these tests are rarely needed in the clinical setting. Patency can also be determined, when clinically indicated, by cholangiography carried out either intraoperatively, percutaneously by transhepatic cholecystography, or by endoscopic retrograde cholangiopancreatography (ERCP) using a pediatric-size side-viewing endoscope. Magnetic resonance cholangiopancreatography in infants is of limited use and highly dependent on the operator and equipment.
1. Perinatal or Neonatal Hepatitis Resulting from Infection
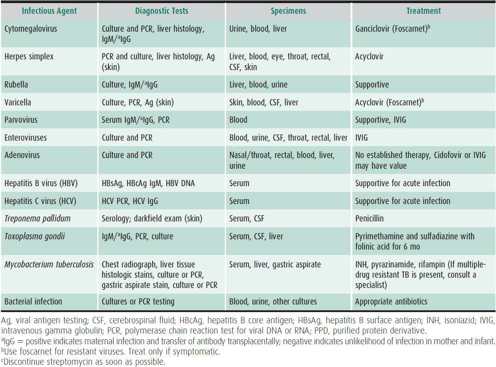
This diagnosis is considered in infants with jaundice, hepatomegaly, vomiting, lethargy, fever, and petechiae. It is important to identify perinatally acquired viral, bacterial, or protozoal infections (Table 22–3). Infection may occur transplacentally, by ascent through the cervix into amniotic fluid, from swallowed contaminated fluids (maternal blood, urine, vaginal secretions) during delivery, from blood transfusions administered in the early neonatal period, or from breast milk or environmental exposure. Infectious agents associated with neonatal intrahepatic cholestasis include herpes simplex virus, varicella virus, enteroviruses (coxsackievirus and echovirus), cytomegalovirus (CMV), rubella virus, adenovirus, parvovirus, human herpesvirus type 6 (HHV-6), hepatitis B virus (HBV), human immunodeficiency virus (HIV), Treponema pallidum, and Toxoplasma gondii. Although hepatitis C may be transmitted vertically, it rarely causes neonatal cholestasis. The degree of liver cell injury caused by these agents is variable, ranging from massive hepatic necrosis (herpes simplex, enteroviruses) to focal necrosis and mild inflammation (CMV, HBV). Serum bilirubin, alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase, and bile acids are typically elevated. The infant is jaundiced, may have petechiae or rash, and generally appears ill.
Table 22–3. Infectious causes of neonatal hepatitis.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Clinical symptoms typically present in the first 2 weeks of life, but may appear as late as age 2–3 months. Jaundice may be noted as early as the first 24 hours of life. Poor oral intake, poor sucking reflex, lethargy, hypotonia, and vomiting are frequent. Stools may be normal to pale in color, but are seldom acholic. Dark urine stains the diaper. Firm hepatomegaly is present and splenomegaly is variably present. Macular, papular, vesicular, or petechial rashes may occur. In less severe cases, failure to thrive may be the primary problem. Unusual presentations include neonatal liver failure, hypoproteinemia, anasarca (nonhemolytic hydrops), and hemorrhagic disease of the newborn.
B. Diagnostic Studies
Neutropenia, thrombocytopenia, and signs of mild hemolysis are common. Mixed hyperbilirubinemia, elevated aminotransferases with near-normal alkaline phosphatase, prolongation of clotting studies, mild acidosis, and elevated cord serum IgM suggest congenital infection. Nasopharyngeal washings, urine, stool, serum, and cerebrospinal fluid (CSF) should be cultured for virus and tested for pathogen-specific nucleic acid. Specific IgM antibody may be useful, as are long-bone radiographs to determine the presence of “celery stalking” in the metaphyseal regions of the humeri, femurs, and tibias. When indicated, computed tomography (CT) scans can identify intracranial calcifications (especially with CMV and toxoplasmosis). Hepatobiliary scintigraphy shows decreased hepatic clearance of the circulating isotope with intact excretion into the gut. Careful ophthalmologic examination may be useful for diagnosis of herpes simplex virus, CMV, toxoplasmosis, and rubella.
A percutaneous liver biopsy is useful in distinguishing intrahepatic from extrahepatic cholestasis, but may not identify a specific infectious agent (see Table 22–2). Exceptions are the typical inclusions of CMV in hepatocytes or bile duct epithelial cells, the presence of intranuclear acidophilic inclusions of herpes simplex or varicella-zoster virus, the presence of adenovirus basophilic intranuclear inclusions, or positive immunohistochemical stains for several viruses. Variable degrees of lobular disarray characterized by focal necrosis, multinucleated giant-cell transformation, and ballooned pale hepatocytes with loss of cordlike arrangement of liver cells are usual. Intrahepatocytic and canalicular cholestasis may be prominent. Portal changes are not striking, but modest neoductular proliferation and mild fibrosis may occur. Viral cultures, immunohistochemical stains, or polymerase chain reaction (PCR) testing of biopsy material may be helpful.
 Differential Diagnosis
Differential Diagnosis
Great care must be taken to distinguish infectious causes of intrahepatic cholestasis from genetic or metabolic disorders because the clinical presentations are similar and may overlap. Galactosemia, hereditary fructose intolerance, and tyrosinemia must be investigated promptly, because specific dietary or drug therapy is available. These infants may also have concomitant bacteremia. α1-antitrypsin deficiency, cystic fibrosis, bile acid synthesis defects, progressive familial intrahepatic cholestasis, mitochondrial respiratory chain disorders, and neonatal iron storage disease must also be considered. Specific physical features may suggest Alagille, arthrogryposis/renal dysfunction/cholestasis (ARC) syndrome or Zellweger syndrome. Idiopathic neonatal hepatitis can be indistinguishable from infectious causes.
Patients with intrahepatic cholestasis frequently appear ill, whereas infants with extrahepatic cholestasis do not typically appear ill, have stools that are usually completely acholic, and have an enlarged, firm liver. Histologic findings are described in Table 22–2.
 Treatment
Treatment
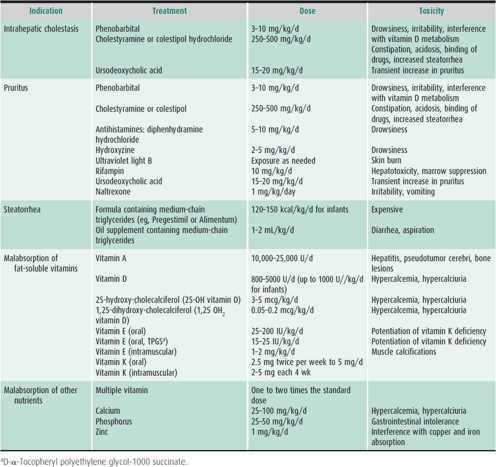
Most forms of viral neonatal hepatitis are treated symptomatically. However, infections with herpes simplex virus, varicella, CMV, parvovirus, and toxoplasmosis have specific treatments (see Table 22–3). Penicillin for suspected syphilis, specific antiviral therapy, or antibiotics for bacterial hepatitis need to be administered promptly. Fluids and adequate nutritional intake are encouraged. Intravenous dextrose is needed if feedings are not well tolerated. The consequences of cholestasis are treated as indicated (Table 22–4). Vitamin K orally or by injection and vitamins D and E orally should be provided. Choleretics (ursodeoxycholic acid [UDCA] or cholestyramine) are used if cholestasis persists. Corticosteroids are contraindicated.
Table 22–4. Treatment of complications of chronic cholestatic liver disease.
 Prognosis
Prognosis
Multiple organ involvement is commonly associated with neonatal infectious hepatitis and has a poor outcome. Hepatic or cardiac failure, intractable acidosis, or intracranial hemorrhage may be fatal in herpesvirus, adenovirus, or enterovirus infections, and occasionally in CMV or rubella infection. HBV rarely causes fulminant neonatal hepatitis; most infected infants are immunotolerant to hepatitis B. Although persistent liver disease with any virus can result in mild chronic hepatitis, portal fibrosis, or cirrhosis, the neonatal liver usually recovers without fibrosis after acute infections. Chronic cholestasis, although rare following infections, may lead to dental enamel hypoplasia, failure to thrive, biliary rickets, severe pruritus, and xanthoma.
Brumbaugh D, Mack C: Conjugated hyperbilirubinemia in children. Pediatr Rev 2012 Jul;33(7):291–302 [PMID: 22753787].
2. Specific Infectious Agents
A. Neonatal Hepatitis B Virus Disease
Vertical transmission of HBV may occur at any time during perinatal life. Most cases of neonatal disease are acquired from mothers who are asymptomatic carriers of HBV. Although HBV has been found in most body fluids, including breast milk, neonatal transmission occurs primarily from exposure to maternal blood at delivery and only occasionally transplacentally (<5%–10% of cases). In chronic hepatitis B surface antigen (HBsAg)–carrier mothers, neonatal acquisition risk is greatest if the mother (1) is also hepatitis B “e” antigen (HBeAg)–positive and hepatitis B “e” antibody (HBeAb)–negative, (2) has high serum levels of hepatitis B core antibody (HBcAb), or (3) has high blood levels of HBV DNA, indicating circulating infectious virus.
Neonatal HBV liver disease is extremely variable. The infant has a 70%–90% chance of acquiring HBV at birth from an HBsAg/HBeAg-positive mother if the infant does not receive prophylaxis. Most infected infants develop a prolonged asymptomatic immunotolerant stage of HBV infection. Fulminant hepatic necrosis and liver failure rarely occur in infants. Other patients develop chronic hepatitis with focal hepatocyte necrosis and a mild portal inflammatory response. Cholestasis is intracellular and canalicular. Chronic hepatitis may persist for many years, with serologic evidence of persisting antigenemia (HBsAg) and mildly elevated or normal serum aminotransferases. Chronic hepatitis may rarely progress to cirrhosis within 1–2 years. Most infected infants have only mild biochemical evidence, if any, of liver injury and do not appear ill. Most infants remain asymptomatic in an immune-tolerant state of HBV infection for a variable period of time and become an inactive carrier, develop chronic hepatitis or remain immune tolerant through childhood (see section on Hepatitis B).
To prevent perinatal transmission, all infants of mothers who are HBsAg-positive (regardless of HBeAg status) should receive hepatitis B immunoglobulin (HBIG) and hepatitis B vaccine within the first 24 hours after birth and vaccine again at ages 1 and 6 months (see Chapter 10). This prevents HBV infection in 85%–95% of infants. HBIG can provide some protection when given as late as 72 hours after birth. If not given at birth it can be administered as late as 7 days postpartum as long as the infant has received the vaccine. Universal HBV immunization at birth, with two follow-up doses, is recommended for all infants regardless of maternal HBV status. Universal screening of pregnant women for HbsAg is conducted to determine which infants will need HBIG.
B. Neonatal Bacterial Hepatitis
Most bacterial liver infections in newborns are acquired by transplacental invasion from amnionitis with ascending spread from maternal vaginal or cervical infection. Onset is abrupt, usually within 48–72 hours after delivery, with signs of sepsis and often shock. Jaundice appears early with direct hyperbilirubinemia. The liver enlarges rapidly, and the histologic picture is that of diffuse hepatitis with or without microabscesses. The most common organisms involved are Escherichia coli, Listeria monocytogenes, and group B streptococci. Neonatal liver abscesses caused by E coli or Staphylococcus aureus may result from omphalitis or umbilical vein catheterization. Bacterial hepatitis and neonatal liver abscesses require specific antibiotics in optimal doses and combinations and, rarely, surgical or radiologic interventional drainage. Deaths are common, but survivors show no long-term consequences of liver disease.
C. Neonatal Jaundice with Urinary Tract Infection
Urinary tract infections typically present with cholestasis between the second and fourth weeks of life. Lethargy, fever, poor appetite, jaundice, and hepatomegaly may be present. Except for mixed hyperbilirubinemia, other liver function tests (LFTs) are mildly abnormal. Leukocytosis is present, and infection is confirmed by urine culture. The liver impairment is caused by the action of endotoxin and cytokines on bile secretion.
Treatment of the infection leads to resolution of the cholestasis without hepatic sequelae. Metabolic liver diseases, such as galactosemia and tyrosinemia, may present with gram-negative bacterial urinary tract infection and must be excluded.
Cherpes TL, Matthews DB, Maryak SA. Neonatal herpes simplex virus infection. Clin Obstet Gynecol 2012;55:938 [PMID: 23090462].
Hendrickx G, Vorsters A, Van Damme P. Advances in hepatitis immunization (A, B, E): public health policy and novel vaccine delivery. Curr Opin Infect Dis 2012;25:578 [PMID: 22907280].
Tran TT: Hepatitis B: treatment to prevent perinatal transmission. Clin Obstet Gynecol 2012;55:541 [PMID: 22510637].
3. Intrahepatic Cholestasis Resulting from Inborn Errors of Metabolism, Familial, & “Toxic” Causes
These cholestatic syndromes caused by specific enzyme deficiencies, other genetic disorders, or certain toxins share findings of intrahepatic cholestasis (ie, jaundice, hepatomegaly, and normal to completely acholic stools). Specific clinical conditions have characteristic clinical signs.
A. Enzyme Deficiencies and Other Inherited Disorders
Establishing the specific diagnosis as early as possible is important because dietary or pharmacologic treatment may be available (Table 22–5). Reversal of liver disease and clinical symptoms may be prompt and maintained in several disorders as long as the diet is maintained. As with other genetic disorders, parents of the affected infant should be offered genetic counseling. For some disorders, prenatal genetic diagnosis is available.
Table 22–5. Metabolic and genetic causes of neonatal cholestasis.
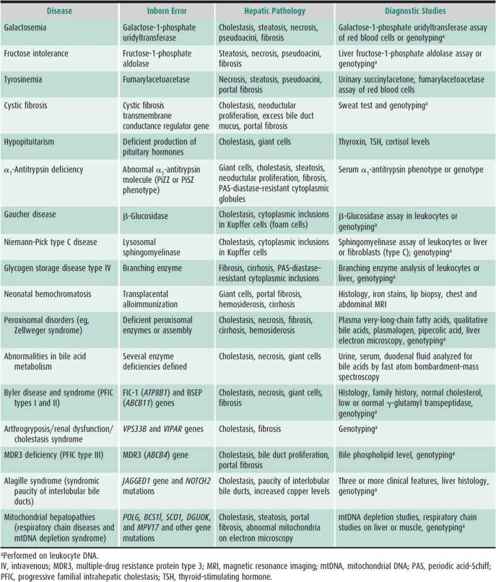
Cholestasis caused by metabolic diseases (eg, galactosemia, hereditary fructose intolerance, and tyrosinemia) is frequently accompanied by vomiting, lethargy, poor feeding, hypoglycemia, or irritability. The infants often appear septic; gram-negative bacteria can be cultured from blood in 25%–50% of symptomatic cases, especially in patients with galactosemia and cholestasis. Neonatal screening programs for galactosemia usually detect the disorder before cholestasis develops. Other metabolic and genetic causes of neonatal intrahepatic cholestasis are outlined in Table 22–5. Treatment of these disorders is discussed in Chapter 36.
B. “Toxic” Causes of Neonatal Cholestasis
1. Neonatal ischemic-hypoxic conditions—Perinatal events that result in hypoperfusion of the gastrointestinal system are sometimes followed within 1–2 weeks by cholestasis. This occurs in preterm infants with respiratory distress, severe hypoxia, hypoglycemia, shock, and acidosis. When these perinatal conditions develop in association with gastrointestinal lesions, such as ruptured omphalocele, gastroschisis, or necrotizing enterocolitis, a subsequent cholestatic picture is common (25%–50% of cases). Liver function studies reveal mixed hyperbilirubinemia, elevated alkaline phosphatase and γ-glutamyl transpeptidase (GGT) values, and variable elevation of the aminotransferases. Stools are seldom persistently acholic.
The mainstays of treatment are choleretics (UDCA), introduction of enteral feedings using special formulas as soon as possible, and nutrient supplementation until the cholestasis resolves (see Table 22–4). As long as no severe intestinal problem is present (eg, short gut syndrome or intestinal failure), resolution of the hepatic abnormalities is the rule, although this may take many weeks.
2. Prolonged parenteral nutrition—Cholestasis may develop after 1–2 weeks in premature newborns receiving parenteral nutrition. Even full-term infants with significant intestinal atresia, resections, or dysmotility may develop parenteral nutrition–associated cholestasis. Contributing factors include toxicity of intravenous lipid emulsions, diminished stimulation of bile flow from prolonged absence of feedings, frequent episodes of sepsis, small intestinal bacterial overgrowth with translocation of intestinal bacteria and their cell wall products, missing nutrients or antioxidants, photooxidation of amino acids, infusion of lipid hydroperoxides or plant sterols, and the “physiologic cholestatic” propensity of the premature infant. Activation of innate immune pathways in the liver appears to be involved. Histology of the liver may be identical to that of biliary atresia. Early introduction of feedings has reduced the frequency of this disorder. The prognosis is generally good; however, in infants with intestinal failure occasional cases progress to cirrhosis, liver failure, and hepatoma. These infants may require liver and intestinal, or multivisceral, transplantation. Oral erythromycin as a pro-motility agent may reduce the incidence of cholestasis in very-low-birth-weight infants. Intravenous fish oil–based lipid emulsions or reduction in soy-oil-based lipid emulsions may reverse features of cholestasis.
3. Inspissated bile syndrome—This syndrome is the result of accumulation of bile in canaliculi and in the small- and medium-sized bile ducts in hemolytic disease of the newborn (Rh, ABO) and in some infants receiving parenteral nutrition. The same mechanisms may cause intrinsic obstruction of the common bile duct. An ischemia-reperfusion injury may also contribute to cholestasis in Rh incompatibility. Stools may become acholic and levels of bilirubin, primarily conjugated, may reach 40 mg/dL. If inspissation of bile occurs within the extrahepatic biliary tree, differentiation from biliary atresia may be difficult. Although most cases improve slowly over 2–6 months, persistence of complete cholestasis for more than 1–2 weeks requires further studies (ultrasonography, hepatobiliary iminodiacetic acid [HIDA] scanning, liver biopsy) with possible cholangiography. Irrigation of the common bile duct is sometimes necessary to dislodge the obstructing inspissated biliary material.
El Kasmi KC et al: Toll-like receptor 4-dependent Kupffer cell activation and liver injury in a novel mouse model of parenteral nutrition and intestinal injury. Hepatology 2012;55:1518 [PMID: 22120983].
Ng PC et al: High-dose oral erythromycin decreased the incidence of parenteral nutrition-associated cholestasis in preterm infants. Gastroenterology 2007;132:1726 [PMID: 17484870].
Rangel SJ et al: Parenteral nutrition-associated cholestasis: an American Pediatric Surgical Association Outcomes and Clinical Trials Committee systematic review. J Pediatr Surg 2012;47:225 [PMID: 22244423].
4. Idiopathic Neonatal Hepatitis (Giant-Cell Hepatitis)
This idiopathic type of cholestatic jaundice, which has a typical liver biopsy appearance, accounts for 20%–30% of cases of neonatal intrahepatic cholestasis but is decreasing in frequency as new causes of cholestasis are discovered. The degree of cholestasis is variable, and the disorder may be indistinguishable from extrahepatic causes in 10% of cases. Many cases of this disorder have been shown in recent years to have specific etiologies. Viral infections, α1-antitrypsin deficiency, Alagille syndrome, Niemann-Pick type C disease (NPC), progressive familial intrahepatic cholestasis (PFIC), citrin deficiency, neonatal hemochromatosis, and bile acid synthesis defects may present with similar clinical and histologic features. In idiopathic neonatal hepatitis, PFIC types I and II and ARC syndrome, and disease due to bile acid synthesis defects, the GGT levels are normal or low. Electron microscopy of the liver biopsy and genotyping will help distinguish NPC and PFIC.
Intrauterine growth retardation, prematurity, poor feeding, emesis, poor growth, and partially or intermittently acholic stools are characteristic of intrahepatic cholestasis. Serious hemorrhage from vitamin K deficiency may also be present. Patients with neonatal lupus erythematosus may present with giant-cell hepatitis; however, thrombocytopenia, rash, or congenital heart block is usually also present.
In cases of suspected idiopathic neonatal hepatitis (diagnosed in the absence of infectious, known genetic, metabolic, and toxic causes), patency of the biliary tree may need to be verified to exclude extrahepatic surgical disorders. HIDA scanning and ultrasonography may be helpful in this regard if stools are acholic. Liver biopsy findings are usually diagnostic after age 6–8 weeks (see Table 22–2), but may be misleading before age 6 weeks. Failure to detect patency of the biliary tree, nondiagnostic liver biopsy findings, or persisting complete cholestasis (acholic stools) are indications for minilaparotomy and intraoperative cholangiography performed by an experienced surgeon, ERCP, percutaneous cholecystography, or magnetic resonance cholangiopancreatography (MRCP). Occasionally, a small but patent (hypoplastic) extrahepatic biliary tree is demonstrated (as in Alagille syndrome); it is probably the result, rather than the cause, of diminished bile flow. Surgical reconstruction of hypoplastic biliary trees in Alagille syndrome should not be attempted.
Therapy should include choleretics, a special formula with medium-chain triglycerides (eg, Pregestimil, Alimentum) or breast milk (if growth is adequate), and supplemental fat-soluble vitamins in water-soluble form (see Table 22–4). This therapy is continued as long as significant cholestasis remains (conjugated bilirubin > 1 mg/dL). Fat-soluble vitamin serum levels and INR should be monitored at regular intervals while supplements are given and repeated at least once after their discontinuation.
Eighty percent of patients recover without significant hepatic fibrosis. However, failure to resolve the cholestatic picture by age 6–12 months is associated with progressive liver disease and evolving cirrhosis, most likely caused by a yet to be defined underlying genetic/metabolic disorder. This may occur with either normal or diminished numbers of interlobular bile ducts (paucity of interlobular ducts). Liver transplantation has been successful when signs of hepatic decompensation are noted (rising bilirubin, coagulopathy, intractable ascites).
Guddat SS et al. Fatal spontaneous subdural bleeding due to neonatal giant cell hepatitis: a rare differential diagnosis of shaken baby syndrome. Forensic Sci Med Pathol 2011 Sep;7(3):294–297 [PMID: 21331818].
Torbenson M et al: Neonatal giant cell hepatitis: histological and etiological findings. Am J Surg Pathol 2010 Oct;34(10): 1498–1503 [PMID: 20871223].
5. Paucity of Interlobular Bile Ducts
Forms of intrahepatic cholestasis caused by decreased numbers of interlobular bile ducts (< 0.5 bile ducts per portal tract) may be classified according to whether they are associated with other malformations. Alagille syndrome (syndromic paucity or arteriohepatic dysplasia) is caused by mutations in the gene JAGGED1, located on chromosome 20p, which codes for a ligand of the notch receptor, or more rarely in the gene NOTCH2. Alagille syndrome is recognized by the characteristic facies, which becomes more obvious with age. The forehead is prominent. The eyes are set deep and sometimes widely apart (hypertelorism). The chin is small and slightly pointed and projects forward. The ears are prominent. The stool color varies with the severity of cholestasis. Pruritus begins by age 3–6 months. Firm, smooth hepatomegaly may be present or the liver may be of normal size. Cardiac murmurs are present in 95% of patients, and butterfly vertebrae (incomplete fusion of the vertebral body or anterior arch) are present in 50%. Xanthomas develop as hypercholesterolemia becomes a problem. Occasionally, early cholestasis is mild and not recognized or the patient presents with complex congenital heart disease (eg, tetralogy of Fallot).
Conjugated hyperbilirubinemia may be mild to severe (2–15 mg/dL). Serum alkaline phosphatase, GGT, and cholesterol are markedly elevated, especially early in life. Serum bile acids are always elevated, aminotransferases are mildly increased, but clotting factors and other liver proteins are usually normal.
Cardiac involvement includes peripheral pulmonary artery, branch pulmonary artery, or pulmonary valvular stenoses, atrial septal defect, coarctation of the aorta, and tetralogy of Fallot. Up to 10%–15% of patients have intracranial vascular or cystic abnormalities or may develop intracranial hemorrhage or stroke early in childhood.
Eye findings (posterior embryotoxon or a prominent Schwalbe line in 90%) and renal abnormalities (dysplastic kidneys, renal tubular ectasia, single kidney, RTA, hematuria) are also characteristic and occur in about 40% of patients. Growth retardation with normal to increased levels of growth hormone (growth hormone resistance) is common. Some patients may rarely have pancreatic insufficiency that may contribute to the fat malabsorption. Although variable, the intelligence quotient is frequently low. Hypogonadism with micropenis may be present. A weak, high-pitched voice may develop. Neurologic disorders resulting from vitamin E deficiency (areflexia, ataxia, ophthalmoplegia) eventually develop in many unsupplemented children and may be profound.
In the nonsyndromic form, paucity of interlobular bile ducts occurs in the absence of the extrahepatic malformations may also occur in conditions such as α1-antitrypsin deficiency, Zellweger syndrome, in association with lymphedema (Aagenaes syndrome), PFIC, cystic fibrosis, CMV or rubella infection, and inborn errors of bile acid metabolism.
High doses (250 mg/kg/d) of cholestyramine may control pruritus, lower cholesterol, and clear xanthomas. UDCA (15–20 mg/kg/d) appears to be more effective and causes fewer side effects than cholestyramine. Rifampicin may also reduce pruritus. Naltrexone (1 mg/kg/d) is occasionally required. Partial biliary diversion or ileal exclusion surgery may reduce pruritus in about half of severe cases. Nutritional therapy to prevent wasting and deficiencies of fat-soluble vitamins is of particular importance because of the severity of cholestasis (see Table 22–4).
Prognosis is more favorable in the syndromic than in the nonsyndromic varieties. In the former, only 30%–40% of patients have severe complications of disease, whereas over 70% of patients with nonsyndromic varieties progress to cirrhosis. Many of this latter group may have forms of PFIC. In Alagille syndrome, cholestasis may improve by age 2–4 years, with minimal residual hepatic fibrosis. Survival into adulthood despite raised serum bile acids, aminotransferases, and alkaline phosphatase occurs in about 50% of cases. Several patients have developed hepatocellular carcinoma. Hypogonadism has been noted; however, fertility is not obviously affected in most cases. Cardiovascular anomalies and intracranial vascular lesions may shorten life expectancy. Some patients have persistent, severe cholestasis, rendering their quality of life poor. Recurrent bone fractures may result from metabolic bone disease. Liver transplantation has been successfully performed under these circumstances. Intracranial hemorrhage, moya moya disease, or stroke may occur in up to 10%–12% of affected children.
Kamath BM et al: Medical management of Alagille syndrome. J Pediatr Gastroenterol Nutr 2010;50:580 [PMID: 20479679].
Kamath BM et al: Outcomes of liver transplantation for patients with Alagille syndrome: the studies of pediatric liver transplantation experience. Liver Transpl 2012;18:940 [PMID: 22454296].
Subramaniam P et al: Diagnosis of Alagille syndrome – 25 years of experience at King’s College Hospital. J Pediatr Gastroenterol Nutr 2011;52:84 [PMID: 21119543].
6. Progressive Familial Intrahepatic Cholestasis (PFIC; Byler Disease and Byler Syndrome)
PFIC is a group of disorders presenting as pruritus, diarrhea, jaundice, fat-soluble vitamin deficiencies, and failure to thrive in the first 6–12 months of life. PFIC type I (Byler disease), caused by mutations in the gene coding FIC1, an aminophospholipid floppase, is associated with low to normal serum levels of GGT and cholesterol and elevated levels of bilirubin, aminotransferases, and bile acids. Pancreatitis and hearing loss may develop. Liver biopsy demonstrates cellular cholestasis, sometimes with a paucity of interlobular bile ducts and centrilobular fibrosis that progresses to cirrhosis. Giant cells are absent. Electron microscopy shows diagnostic granular “Byler bile” in canaliculi. Treatment includes administration of UDCA, partial biliary diversion or ileal exclusion if the condition is unresponsive to UDCA, and liver transplantation if unresponsive to these therapies. With partial biliary diversion or ileal exclusion surgery, many patients show improved growth and liver histology, reduction in symptoms and, thus, avoid liver transplantation. Following liver transplantation, chronic diarrhea and fatty liver may complicate recovery.
PFIC type II is caused by mutations in the bile salt export pump (BSEP) gene, which codes for the adenosine triphosphate–dependent canalicular bile salt transport protein. These patients are clinically and biochemically similar to PFIC type I patients, but liver histology includes numerous “giant cells.” There is an increased incidence of hepatocellular carcinoma in these patients with severe gene mutations. Treatment is similar to PFIC type I although close monitoring for hepatocellular carcinoma is essential. Following liver transplantation, recurrent disease has been described in patients who developed immune-mediated BSEP dysfunction.
PFIC type III is caused by mutations in the multiple drug resistance protein type 3 (MDR3) gene, which encodes a canalicular protein that pumps phospholipid into bile. Serum GGT and bile acid levels are both elevated, bile duct proliferation and portal tract fibrosis are seen in liver biopsies (resembling biliary atresia), and bile phospholipid levels are low. Treatment is similar to that for other forms of PFIC except that partial biliary diversion is not recommended.
Bile acid synthesis defects are clinically similar to PFIC types I and II, with low serum levels of GGT and cholesterol; however, the serum level of total bile acids is inappropriately normal or low and urine bile acid analysis may identify a synthesis defect. Treatment of bile acid synthesis defects is with oral cholic acid and UDCA. About 1/3 of PFIC patients have negative genotyping for the above genes and most likely have yet-to-be discovered genetic etiologies.
Arnell H et al: Follow-up in children with progressive familial intrahepatic cholestasis after partial external biliary diversion. J Pediatr Gastroenterol Nutr 2010;51:494 [PMID: 20683202].
Morotti RA et al: Progressive familial intrahepatic cholestasis (PFIC) type 1, 2, and 3: a review of the liver pathology findings. Semin Liver Dis 2011;31:3 [PMID: 21344347].
van der Woerd WL, van Mil SW, Stapelbroek JM, Klomp LW, van de Graaf SF, Houwen RH. Familial cholestasis: progressive familial intrahepatic cholestasis, benign recurrent intrahepatic cholestasis and intrahepatic cholestasis of pregnancy. Best Pract Res Clin Gastroenterol 2010;24:541 [PMID: 20955958].
EXTRAHEPATIC NEONATAL CHOLESTASIS
Extrahepatic neonatal cholestasis is characterized by complete and persistent cholestasis (acholic stools) in the first 3 months of life; lack of patency of the extrahepatic biliary tree proved by intraoperative, percutaneous, or endoscopic cholangiography; firm to hard hepatomegaly; and typical features on histologic examination of liver biopsy tissue (see Table 22–2). Causes include biliary atresia, choledochal cyst, spontaneous perforation of the extrahepatic ducts, and intrinsic or extrinsic obstruction of the common duct.
1. Biliary Atresia
 General Considerations
General Considerations
Biliary atresia is the progressive fibroinflammatory obliteration of the lumen of all, or part of, the extrahepatic biliary tree presenting within the first 3 months of life. Biliary atresia occurs in 1:6600 (Taiwan)–1:18,000 (Europe) births, and in the United States the incidence is approximately 1:12,000. The incidence is highest in Asians, African Americans, and preterm infants, and there is a slight female predominance. There are at least two types of biliary atresia: the perinatal form (80% of cases), in which a perinatal insult, such as a virus infection, is believed to initiate inflammatory obstruction and fibrosis of the biliary tree, and the fetal-embryonic form (20% of cases), in which the extrahepatic biliary system did not develop normally. In the perinatal form, meconium and initial stools are usually normal in color, suggesting early patency of the ducts. Evidence obtained from surgically removed remnants of the extrahepatic biliary tree suggests an inflammatory sclerosing cholangiopathy. Recent research supports an autoimmune reaction that is responsible for progressive intrahepatic bile duct injury and fibrosis. In the fetal-embryonic type, the bile duct presumably did not develop normally and is associated with other nonhepatic congenital anomalies. The association of biliary atresia with the polysplenia syndrome (heterotaxia, preduodenal portal vein, interruption of the inferior vena cava, polysplenia, and midline liver) and asplenia syndrome supports an embryonic origin of biliary atresia in these cases.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Jaundice may be noted in the newborn period or develops about age 2–3 weeks. Urine stains the diaper; and stools are often pale yellow, buff-colored, gray, or acholic. Seepage of bilirubin products across the intestinal mucosa may give some yellow coloration to the stools. Hepatomegaly is common, and the liver may feel firm to hard. By age 2–6 months, the growth curve reveals poor weight gain. Symptoms of portal hypertension (splenomegaly, ascites, variceal bleeding) may develop in the first year of life. Pruritus, digital clubbing, failure to thrive, bone fractures, and bleeding complications may also occur later in childhood.
B. Laboratory Findings and Imaging
No single laboratory test will consistently differentiate biliary atresia from other causes of complete obstructive jaundice. Although biliary atresia is suggested by persistent elevation of serum GGT or alkaline phosphatase levels, these findings have also been reported in severe neonatal hepatitis, α1-antitrypsin deficiency, and bile duct paucity. Furthermore, these tests will not differentiate the location of the obstruction within the extrahepatic system. Generally, the aminotransferases are only modestly elevated in biliary atresia. Serum proteins and blood clotting factors are not affected early in the disease. Ultrasonography of the biliary system should be performed to exclude the presence of choledochal cyst and intra-abdominal anomalies; a triangular cord sign in the hepatic porta suggests biliary atresia. A HIDA scan showing lack of intestinal excretion is always present in biliary atresia, but may be seen with multiple other causes of intrahepatic cholestasis. Liver biopsy specimens (particularly if obtained after age 6–8 weeks) can differentiate intrahepatic causes of cholestasis from biliary atresia in over 90% of cases (see Table 22–2).
 Differential Diagnosis
Differential Diagnosis
The major diagnostic dilemma is distinguishing between this entity and bile duct paucity, metabolic liver disease (particularly α1-antitrypsin deficiency), choledochal cyst, or intrinsic bile duct obstruction (stones, bile plugs). Although spontaneous perforation of extrahepatic bile ducts leads to jaundice and acholic stools, the infants in such cases are usually quite ill with chemical peritonitis from biliary ascites, and hepatomegaly is not found.
If the diagnosis of biliary atresia cannot be excluded by the diagnostic evaluation and percutaneous liver biopsy, surgical exploration should be performed as soon as possible. Laparotomy or laparoscopy must include liver biopsy and an operative cholangiogram if a gallbladder is present. The presence of yellow bile in the gallbladder implies patency of the proximal extrahepatic duct system. Radiographic visualization of cholangiographic contrast in the duodenum excludes obstruction to the distal extrahepatic ducts.
 Treatment
Treatment
In the absence of surgical correction or transplantation, biliary cirrhosis, hepatic failure, and death occur uniformly by age 18–24 months.
The standard procedure at the time of diagnosis of biliary atresia is the hepatoportoenterostomy (Kasai procedure). Occasionally, portocholecystostomy (gallbladder Kasai procedure) may be performed if the gallbladder is present and the passage from it to the duodenum is patent. These procedures are best done in specialized centers where experienced surgical, pediatric, and nursing personnel are available. Surgery should be performed as early as possible (ideally before 45 days of life); the Kasai procedure should generally not be undertaken in infants older than age 4 months, because the likelihood of bile drainage at this age is very low. Orthotopic liver transplantation is now indicated for patients who do not undergo the Kasai procedure, who fail to drain bile after the Kasai procedure, or who progress to end-stage biliary cirrhosis despite surgical treatment. The 3- to 5-year survival rate following liver transplantation for biliary atresia is at least 80%–90%. Biliary atresia is the leading indication for pediatric liver transplantation.
Whether or not the Kasai procedure is performed, supportive medical treatment consists of vitamin and caloric support (vitamins A, D, E, and K and formulas containing medium-chain triglycerides [Pregestimil or Alimentum]) (see Table 22–4). Monitoring of fat-soluble vitamin levels is essential to ensure adequate supplementation. Suspected bacterial infections (eg, ascending cholangitis) should be treated promptly with broad-spectrum antibiotics, and any bleeding tendency should be corrected with intramuscular vitamin K. Ascites can be managed initially with reduced sodium intake and spironolactone. Choleretics and bile acid–binding products (cholestyramine, aluminum hydroxide gel) are of little use. The value of UDCA remains to be determined. Antibiotic prophylaxis reduces the recurrence rate of cholangitis. The role of post-Kasai corticosteroids is controversial.
 Prognosis
Prognosis
When bile flow is sustained following portoenterostomy (serum total bilirubin < 2 mg/dL by 3 months of age), the 10-year survival rate without liver transplantation is up to 35%. Death is usually caused by liver failure, sepsis, intractable variceal bleeding or respiratory failure secondary to intractable ascites. Esophageal variceal hemorrhage develops in 40% of patients, yet terminal hemorrhage is unusual. Occasional long-term survivors develop hepatopulmonary syndrome (intrapulmonary right to left shunting of blood resulting in hypoxia) or portopulmonary hypertension (pulmonary arterial hypertension in patients with portal hypertension). Liver transplantation has dramatically changed the outlook for these patients.
Chiu CY et al: Taiwan infant stool color card study group. Biliary atresia in preterm infants in Taiwan: a nationwide survey. J Pediatr 2013 epub ahead of print [PMID: 23414661].
Mack CL et al: Clues to the etiology of bile duct injury in biliary atresia. Semin Liver Dis 2012;32:307 [PMID: 23414661].
Superina R et al: The anatomic pattern of biliary atresia identified at time of Kasai hepatoportoenterostomy and early postoperative clearance of jaundice are significant predictors of transplant-free survival. Ann Surg 2011;254:577 [PMID: 21869674].
2. Choledochal Cyst
 ESSENTIALS OF DIAGNOSIS
ESSENTIALS OF DIAGNOSIS
 Abnormal abdominal ultrasound with cyst of the biliary tree.
Abnormal abdominal ultrasound with cyst of the biliary tree.
 Clinical Features
Clinical Features
A. Symptoms and Signs
Choledochal cysts are cystic lesions of all or part of the extrahepatic biliary system and in rare cases the cystic malformation can include the intrahepatic bile duct branches. In most cases, the clinical manifestations, basic laboratory findings, and histopathologic features on liver biopsy are indistinguishable from those associated with biliary atresia. In older children, choledochal cyst presents as recurrent episodes of right upper quadrant abdominal pain, fevers, vomiting, obstructive jaundice, pancreatitis, or as a right abdominal mass. Infants and children with choledochal cysts are at increased risk for developing bacterial cholangitis. Choledochal cysts cause only 2%–5% of cases of extrahepatic neonatal cholestasis; the incidence is higher in girls and patients of Asian descent. Neonatal symptomatic cysts may be associated with atresia of the distal common duct—accounting for the diagnostic dilemma—and may simply be part of the spectrum of biliary atresia.
B. Imaging Studies
Ultrasonography or magnetic resonance imaging (MRI) reveals the presence of a cyst.
 Treatment
Treatment
Timely surgery is indicated for neonates once abnormalities in clotting factors have been corrected and bacterial cholangitis, if present, has been treated with intravenous antibiotics. Excision of the cyst and choledocho–Roux-en-Y jejunal anastomosis are recommended. In some cases, because of technical problems, only the mucosa of the cyst can be removed with jejunal anastomosis to the proximal bile duct. Anastomosis of cyst to jejunum or duodenum is not recommended due to the continued risks of cholangitis and bile duct carcinoma (cholangiocarcinoma).
 Prognosis
Prognosis
The prognosis depends on the presence or absence of associated evidence of atresia and the appearance of the intrahepatic ducts. If atresia is found, the prognosis is similar to that described in the preceding section. If an isolated extrahepatic cyst is encountered, the outcome is generally excellent, with resolution of the jaundice and return to normal liver architecture. However, bouts of ascending cholangitis may occur, particularly if intrahepatic cysts are present or stricture of the anastomotic site develops. The risk of cholangiocarcinoma developing within the cyst is about 5%–15% in adulthood; therefore, cystectomy or excision of cyst mucosa should be undertaken whenever possible.
Hung MH et al: Choledochal cysts in infants and children: experiences over a 20-year period at a single institution. Eur J Pediatr 2011;170:1179 [PMID: 21350805].
Tsai MS et al: Clinicopathological feature and surgical outcome of choledochal cyst in different age groups: the implication of surgical timing. J Gastrointest Surg 2008;12:2191 [PMID: 18677540].
3. Spontaneous Perforation of the Extrahepatic Bile Ducts
The sudden appearance of obstructive jaundice, acholic stools, and abdominal enlargement with ascites in a sick newborn is suggestive of this condition. The liver is usually normal in size, and a yellow-green discoloration can often be discerned under the umbilicus or in the scrotum. In 24% of cases, stones or sludge obstructs the common bile duct. HIDA scan or ERCP shows leakage from the biliary tree, and ultrasonography confirms ascites or fluid around the bile duct.
Treatment is surgical. Simple drainage, without attempts at oversewing the perforation, is sufficient in primary perforations. A diversion anastomosis is constructed in cases associated with choledochal cyst or stenosis. The prognosis is generally good.
Pereira E et al: Conservative management of spontaneous bile duct perforation in infancy: case report and literature review. J Pediatr Surg 2012;47:1757 [PMID: 22974619].
OTHER NEONATAL HYPERBILIRUBINEMIC CONDITIONS (NONCHOLESTATIC NONHEMOLYTIC)
Two other groups of disorders are associated with hyperbilirubinemia: (1) unconjugated hyperbilirubinemia, present in breast milk jaundice, Lucey-Driscoll syndrome, congenital hypothyroidism, upper intestinal obstruction, Gilbert disease, Crigler-Najjar syndrome, and drug-induced hyperbilirubinemia; and (2) conjugated noncholestatic hyperbilirubinemia, present in the Dubin-Johnson syndrome and Rotor syndrome.
1. Unconjugated Hyperbilirubinemia
A. Breast Milk Jaundice
Persistent elevation of the indirect bilirubin fraction (> 80% of total bilirubin) may occur in up to 36% of breast-fed infants. Enhanced β-glucuronidase activity in breast milk is one factor that increases absorption of unconjugated bilirubin. Substances (eg, L-aspartic acid) in casein hydrolysate formulas inhibit this enzyme. The increased enterohepatic shunting of unconjugated bilirubin exceeds the normal conjugating capacity in the liver of these infants. The mutation for Gilbert syndrome (UDP-glucuronyltransferase 1A1 [UGT1A1]) predisposes to breast milk jaundice and to more prolonged jaundice. Neonates who carry the 211 and 388 variants in the UGT1A1 and OATP 2 genes, respectively, and feed with breast milk, are at high risk to develop severe hyperbilirubinemia. Low volumes of ingested breast milk may also contribute to jaundice in the first week of life. Finally, breast milk may suppress UGT1A1 expression in the infant’s intestines which may also lead to unconjugated hyperbilirubinemia.
Hyperbilirubinemia does not usually exceed 20 mg/dL, with most cases in the range of 10–15 mg/dL. Jaundice is noticeable by the fifth to seventh day of breast feeding. It may accentuate the underlying physiologic jaundice—especially early, when total fluid intake may be less than optimal. Except for jaundice, the physical examination is normal; urine does not stain the diaper, and the stools are golden yellow.
The jaundice peaks before the third week of life and clears before age 3 months in almost all infants, even when breast feeding is continued. All infants who remain jaundiced past age 2–3 weeks should have measurement of conjugated bilirubin to exclude cholestasis and hepatobiliary disease.
Kernicterus has rarely been reported in association with this condition. In special situations, breast feeding may be discontinued temporarily and replaced by formula feedings for 2–3 days until serum bilirubin decreases by 2–8 mg/dL. Cow’s milk formulas inhibit the intestinal reabsorption of unconjugated bilirubin. When breast feeding is reinstituted, the serum bilirubin may increase slightly, but not to the previous level. Phototherapy is not indicated in the healthy full-term infant with this condition unless bilirubin levels meet high-risk levels as defined by the American Academy of Pediatrics.
Bhutani VK et al: Expert Committee for Severe Neonatal Hyperbilirubinemia; European Society for Pediatric Research; American Academy of Pediatrics: management of jaundice and prevention of severe neonatal hyperbilirubinemia in infants ≥ 35 weeks gestation. Neonatology 2008;94:63 [PMID: 18204221].
Fujiwara R et al: Reduced expression of UGT1A1 in intestines of humanized UGT1 mice via inactivation of NF-κB leads to hyperbilirubinemia. Gastroenterology 2012;142:109 [PMID: 21983082].
Preer GL, Philipp BL: Understanding and managing breast milk jaundice. Arch Dis Child Fetal Neonatal Ed 2011;96:F461 [PMID: 20688866].
B. Congenital Hypothyroidism
Although the differential diagnosis of indirect hyperbilirubinemia should always include congenital hypothyroidism, the diagnosis is usually suggested by clinical and physical clues or, more commonly, from the newborn screening results. The jaundice clears quickly with replacement thyroid hormone therapy, although the mechanism is unclear.
Tiker F: Congenital hypothyroidism and early severe hyperbilirubinemia. Clin Pediatr (Phila) 2003;42:365 [PMID: 12800733].
C. Upper Intestinal Obstruction
The association of indirect hyperbilirubinemia with high intestinal obstruction (eg, duodenal atresia, annular pancreas, pyloric stenosis) in the newborn has been observed repeatedly; the mechanism is unknown. Diminished levels of hepatic glucuronyl transferase are found on liver biopsy in pyloric stenosis, and genetic studies suggest that this indirect hyperbilirubinemia may be an early sign of Gilbert syndrome.
Treatment is that of the underlying obstructive condition (usually surgical). Jaundice disappears once adequate nutrition is achieved.
Hua L et al: The role of UGT1A1∗28 mutation in jaundiced infants with hypertrophic pyloric stenosis. Pediatr Res 2005;58:881 [PMID: 16257926].
D. Gilbert Syndrome
Gilbert syndrome is a common form of familial hyperbilirubinemia present in 3%–7% of the population. It is associated with a partial reduction of hepatic bilirubin uridine diphosphate-glucuronyl transferase activity. Affected infants may have more rapid increase in jaundice in the newborn period, accentuated breast milk jaundice, and jaundice with intestinal obstruction. During puberty and beyond, mild fluctuating jaundice, especially with illness and vague constitutional symptoms, is common. Shortened red blood cell survival in some patients is thought to be caused by reduced activity of enzymes involved in heme biosynthesis (protoporphyrinogen oxidase). Reduction of hyperbilirubinemia has been achieved in patients by administration of phenobarbital (5–8 mg/kg/d), although this therapy is not justified.
The disease is inherited as an abnormality of the promoter region of uridine diphosphate-glucuronyl transferase-1 (UDGT1); however, another factor appears to be necessary for disease expression. The homozygous (16%) and heterozygous states (40%) are common. Males are affected more often than females (4:1) for reasons that are not clear. Serum unconjugated bilirubin is generally less than 3–6 mg/dL, although unusual cases may exceed 8 mg/dL. The findings on liver biopsy and most LFTs are normal. An increase of 1.4 mg/dL or more in the level of unconjugated bilirubin after a 2-day fast (300 kcal/d) is consistent with the diagnosis of Gilbert syndrome. Gilbert syndrome, conferred by the donor liver, can occur following liver transplantation. Genetic testing is available but rarely needed. No treatment is necessary.
Bartlett MG, Gourley GR: Assessment of UGT polymorphisms and neonatal jaundice. Semin Perinatol 2011;35:127 [PMID: 21641485].
Claridge LC et al: Gilbert’s syndrome. BMJ 2011 Apr 19;342:d2293 [PMID: 21508045].
Kathemann S et al: Gilbert’s syndrome—a frequent cause of unconjugated hyperbilirubinemia in children after orthotopic liver transplantation. Pediatr Transplant 2012;16:20 [PMID: 22360405].
E. Crigler-Najjar Syndrome
Infants with type 1 Crigler-Najjar syndrome usually develop rapid severe unconjugated hyperbilirubinemia (> 30–40 mg/dL) with neurologic consequences (kernicterus). Consanguinity is often present. Prompt recognition of this entity and treatment with exchange transfusions are required, followed by phototherapy. Some patients have no neurologic signs until adolescence or early adulthood, at which time deterioration may occur suddenly. For diagnosis of this condition, it may be useful to obtain a duodenal bile specimen, which characteristically will be colorless and contain a predominance of unconjugated bilirubin, small amounts of monoconjugates, and only traces of unconjugated bilirubin. Phenobarbital administration does not significantly alter these findings, nor does it lower serum bilirubin levels. UGT genetic testing is available. The deficiency in UGT1A1 is inherited in an autosomal recessive pattern. A combination of aggressive phototherapy and cholestyramine may keep bilirubin levels below 25 mg/dL. The use of tin protoporphyrin or tin mesoporphyrin remains experimental. Orlistat therapy may decrease bilirubin in a subset of patients. Liver transplantation is curative and may prevent kernicterus if performed early. Auxiliary orthotopic transplantation also relieves the jaundice while the patient retains native liver. Hepatocyte transplantation is experimental and future gene therapy may be possible
A milder form (type 2) with both autosomal dominant and recessive inheritance is rarely associated with neurologic complications. Hyperbilirubinemia is less severe, and the bile is pigmented and contains small amounts of bilirubin monoglucuronide and diglucuronide. Patients with this form respond to phenobarbital (4 mg/kg/d in infants) with lowering of serum bilirubin levels. An increased proportion of monoconjugated and diconjugated bilirubin in the bile follows phenobarbital treatment. Liver biopsy findings and LFTs are consistently normal in both types.
Bartlett MG, Gourley GR: Assessment of UGT polymorphisms and neonatal jaundice. Semin Perinatol 2011;35:127 [PMID: 21641485].
Kohli S et al: Novel human pathological mutations. Gene symbol: UGT1A1 Disease: Crigler-Najjar syndrome 1. Hum Genet 2010;127:485 [PMID: 21488310].
F. Drug-Induced Hyperbilirubinemia
Vitamin K3 (menadiol) may elevate indirect bilirubin levels by causing hemolysis. Vitamin K1 (phytonadione) can be used safely in neonates. Carbamazepine can cause conjugated hyperbilirubinemia in infancy. Rifampin and antiretroviral protease inhibitors may cause unconjugated hyperbilirubinemia. Pancuronium bromide and chloral hydrate have been implicated in causing neonatal jaundice. Other drugs (eg, ceftriaxone, sulfonamides) may displace bilirubin from albumin, potentially increasing the risk of kernicterus—especially in the sick premature infant.
2. Conjugated Noncholestatic Hyperbilirubinemia (Dubin-Johnson Syndrome & Rotor Syndrome)
These diagnoses are suspected when persistent or recurrent conjugated hyperbilirubinemia and jaundice occur and liver function tests are normal. The basic defect in Dubin-Johnson syndrome is in the multiple organic anion transport protein 2 (MRP2) of the bile canaliculus, causing impaired hepatocyte excretion of conjugated bilirubin into bile. A variable degree of impairment in uptake and conjugation complicates the clinical picture. Transmission is autosomal recessive, so a positive family history is occasionally obtained. In Rotor syndrome, the defect lies in hepatic uptake and storage of bilirubin. OATP1B1 and OATP1B3 are the two transporters that are deficient. Bile acids are metabolized normally, so that cholestasis does not occur. Bilirubin values range from 2 to 5 mg/dL, and other LFTs are normal.
In Rotor syndrome, the liver is normal; in Dubin-Johnson syndrome, it is darkly pigmented on gross inspection and may be enlarged. Microscopic examination reveals numerous dark-brown pigment granules consisting of polymers of epinephrine metabolites, especially in the centrilobular regions. However, the amount of pigment varies within families, and some jaundiced family members may have no demonstrable pigmentation in the liver. Otherwise, the liver is histologically normal. Oral cholecystography fails to visualize the gall-bladder in Dubin-Johnson syndrome, but is normal in Rotor syndrome. Differences in the excretion patterns of bromosulfophthalein, in results of HIDA cholescintigraphy, in urinary coproporphyrin I and III levels, and in the serum pattern of monoglucuronide and diglucuronide conjugates of bilirubin can help distinguish between these two conditions. Genotyping of MRP2 is available but for the Rotor syndrome this is only available on a research basis.
Choleretic agents (eg, UDCA) may help reduce the cholestasis in infants with Dubin-Johnson syndrome.
Jirsa M et al: Rotor syndrome. In: Pagon RA et al (eds): GeneReviews™ [Internet]. 2012 Dec 13 [PMID: 23236639].
Strassburg CP: Hyperbilirubinemia syndromes (Gilbert-Meulengracht, Crigler-Najjar, Dubin-Johnson, and Rotor syndrome). Best Pract Res Clin Gastroenterol 2010;24:555 [PMID: 20955959].
HEPATITIS A
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Gastrointestinal upset (anorexia, vomiting, diarrhea).
Gastrointestinal upset (anorexia, vomiting, diarrhea).
 Jaundice.
Jaundice.
 Liver tenderness and enlargement.
Liver tenderness and enlargement.
 Abnormal LFTs.
Abnormal LFTs.
 Local epidemic of hepatitis A infection.
Local epidemic of hepatitis A infection.
 Positive anti–hepatitis A virus (HAV) IgM antibody.
Positive anti–hepatitis A virus (HAV) IgM antibody.
 Pathogenesis
Pathogenesis
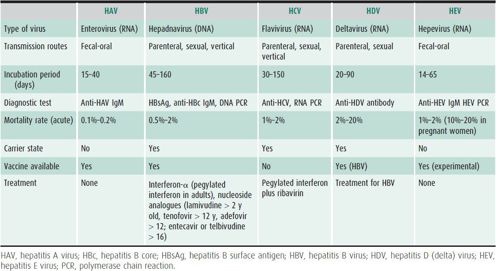
Hepatitis A virus (HAV) infection occurs in both epidemic and sporadic fashion (Table 22–6). Fecal-oral route is the mode of transmission in epidemic outbreaks from contaminated food or water supplies, including by food handlers. HAV viral particles are found in stools during the acute phase of hepatitis A infection. Sporadic cases usually result from contact with an infected individual. Transmission through blood products obtained during the viremic phase is a rare event, although it has occurred in a newborn nursery.
Table 22–6. Hepatitis viruses.
 Prevention
Prevention
Isolation of the patient during initial phases of illness is indicated, although most patients with hepatitis A are noninfectious by the time the disease becomes overt. Stool, diapers, and other fecally stained clothing should be handled with care for 1 week after the appearance of jaundice.
Passive-active immunization of exposed susceptible persons < 12 months old; over 40 years of age: anyone who is immunocompromised or who has chronic liver disease is recommended with immune globulin, 0.02 mL/kg intramuscularly. Illness is prevented in > 85% of individuals if immune globulin is given within 2 weeks of exposure. For individuals 12 months to 40 years old HAV vaccine is recommended following exposure. Infants < 12 months old traveling to endemic disease areas should receive HAV vaccine or 0.02 or 0.06 mL/kg (for trip > 3 months) of immune globulin as prophylaxis. Older individuals should receive the HAV vaccine. All children older than 12 months with chronic liver disease should receive two doses of HAV vaccine 6 months apart. It is currently recommended that all children 12–18 months of age receive HAV vaccination in the United States. If an emigrant child from an endemic area is adopted, the immediate family members should be immunized. Lifelong immunity to HAV follows infection.
Antibody to HAV appears within 1–4 weeks of clinical symptoms. Although the great majority of children with infectious hepatitis are asymptomatic or have mild disease and recover completely, some will develop fulminant hepatitis leading to death or requiring liver transplantation.
 Clinical Findings
Clinical Findings
A. History
Historical risk factors may include direct exposure to a previously jaundiced individual or recently arrived individual from a high prevalence country, consumption of seafood, contaminated water or imported fruits or vegetables, attendance in a day care center, or recent travel to an area of endemic infection. Following an incubation period of 15–40 days, nonspecific symptoms usually precede the development of jaundice by 5–10 days. In developing countries, hepatitis A is common and most children are exposed to HAV by age 10 years, while only 20% are exposed by age 20 years in developed countries.
B. Symptoms and Signs
The overt form of the disease is easily recognized by the clinical manifestations. However, two-thirds of children are asymptomatic, and two-thirds of symptomatic children are anicteric. Therefore, the presenting symptoms in children with HAV resemble gastroenteritis. Fever, anorexia, vomiting, headache, and abdominal pain are typical and dark urine precedes jaundice, which peaks in 1–2 weeks and then begins to subside. The stools may become light- or clay-colored. Clinical improvement can occur as jaundice develops. Tender hepatomegaly and jaundice are typically present; splenomegaly is variable.
C. Laboratory Findings
Serum aminotransferases and conjugated and unconjugated bilirubin levels are elevated. Although unusual, hypoalbuminemia, hypoglycemia, and marked prolongation of PT (international normalized ratio [INR] > 2.0) are serious prognostic findings. Diagnosis is made by a positive anti-HAV IgM, whereas anti-HAV IgG persists after recovery.
Percutaneous liver biopsy is rarely indicated. “Balloon cells” and acidophilic bodies are characteristic histologic findings. Liver cell necrosis may be diffuse or focal, with accompanying infiltration of inflammatory cells containing polymorphonuclear leukocytes, lymphocytes, macrophages, and plasma cells, particularly in portal areas. Some bile duct proliferation may be seen in the perilobular portal areas alongside areas of bile stasis. Regenerative liver cells and proliferation of reticuloendothelial cells are present. Occasionally massive hepatocyte necrosis portends a poor prognosis.
 Differential Diagnosis
Differential Diagnosis
Before jaundice appears, the symptoms are those of nonspecific viral enteritis. Other diseases with somewhat similar onset include pancreatitis, infectious mononucleosis, leptospirosis, drug-induced hepatitis, Wilson disease, autoimmune hepatitis (AIH), and infection with other hepatitis viruses. Acquired CMV disease may also mimic HAV, although lymphadenopathy is usually present in the former.
 Treatment
Treatment
No specific treatment measures are required although bed rest is reasonable for the child who appears ill. Sedatives and corticosteroids should be avoided. During the icteric phase, lower-fat foods may diminish gastrointestinal symptoms, but do not affect overall outcome. Drugs and elective surgery should be avoided. Hospitalization is recommended for children with coagulopathy, encephalopathy, or severe vomiting.
 Prognosis
Prognosis
Ninety-nine percent of children recover without sequelae. Persons with underlying chronic liver disease have an increased risk of death. In rare cases of acute liver failure due to HAV hepatitis, the patient may die within days to weeks and requires evaluation for liver transplantation. The prognosis is poor if hepatic coma or ascites develop; liver transplantation is indicated under these circumstances and is life-saving. Incomplete resolution can cause a prolonged hepatitis; however, resolution invariably occurs without long-term hepatic sequelae. Rare cases of aplastic anemia following acute infectious hepatitis have been reported. A benign relapse of symptoms may occur in 10%–15% of cases after 6–10 weeks of apparent resolution.
Dorell CG et al: Hepatitis A vaccination coverage among adolescents in the United States. Pediatrics 2012;129:213 [PMID: 22271690].
Erhart LM, Ernst KC: The changing epidemiology of hepatitis A in Arizona following intensive immunization programs (1988–2007). Vaccine 2012;30:6103 [PMID: 22835739].
Hepatitis A in Red Book: 2012 report of the committee on infectious diseases, 29th ed, Elk Grove Village, IL. American Academy of Pediatrics; 2012.
Marshall H et al: Long-term (5 year) antibody persistence following two- and three-dose regimens of a combined hepatitis A and B vaccine in children aged 1–11 years. Vaccine 2010;17:4411 [PMID: 20434544].
HEPATITIS B
 ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
ESSENTIALS OF DIAGNOSIS & TYPICAL FEATURES
 Gastrointestinal upset, anorexia, vomiting, diarrhea.
Gastrointestinal upset, anorexia, vomiting, diarrhea.
 Jaundice, tender hepatomegaly, abnormal LFTs.
Jaundice, tender hepatomegaly, abnormal LFTs.
 Serologic evidence of hepatitis B disease: HBsAg, HBeAg, anti-HBc IgM.
Serologic evidence of hepatitis B disease: HBsAg, HBeAg, anti-HBc IgM.
 History of parenteral, sexual, or household exposure or maternal HBsAg carriage.
History of parenteral, sexual, or household exposure or maternal HBsAg carriage.
 General Considerations
General Considerations
In contrast to HAV, hepatitis B virus (HBV) infection has a longer incubation period of 45–160 days (see Table 22–6). HBV is a DNA virus that is either acquired perinatally from a carrier mother, or later in life from exposure to contaminated blood through shared needles, needle sticks, skin piercing, tattoos, or sexual transmission. Transmission via blood products has been almost eliminated by donor-screening and donor blood testing protocols.
 Pathophysiology
Pathophysiology
The HBV particle is composed of a core that is found in the nucleus of infected liver cells and a double outer shell (surface antigen). The surface antigen in blood is termed HBsAg, which elicits an antibody (anti-HBs). The core antigen is termed HBcAg and its antibody is anti-HBc. Anti-HBc IgM antibody indicates recent viral infection. Another important antigen-antibody system associated with HBV disease is the “e” (envelope) antigen system. HBeAg, a truncated soluble form of HBcAg, correlates with active virus replication. Persistence of HBeAg is a marker of infectivity, whereas the appearance of anti-HBe generally implies a lower level of viral replication. However, HBV mutant viruses (precore mutant) may replicate with negative HBeAg tests and positive tests for anti-HBe antibody (HBeAg-negative chronic hepatitis) and are associated with a more virulent form of hepatitis. Circulating HBV DNA (measured by PCR) also indicates viral replication.
 Prevention
Prevention
HBV vaccination is the preferred method for prevention. Universal immunization of all infants born in the United States and of adolescents is now recommended, as it is in most other countries. Other control methods include screening of blood donors and pregnant women, use of properly sterilized needles and surgical equipment, avoidance of sexual contact with carriers, general adoption of safe sex practices, and vaccination of household contacts, sexual partners, medical personnel, and those at high risk. For postexposure prophylaxis, HBV vaccine alone (see Chapter 10) or together with administration of hepatitis B immune globulin (HBIG) (0.06 mL/kg intramuscularly, given as soon as possible after exposure, up to 7 days). The risk of vertical transmission is dramatically reduced with the combination of newborn vaccination and HBIG administration.
 Clinical Findings
Clinical Findings
A. Symptoms and Signs
Most infants and young children are completely asymptomatic, especially if the infection is acquired vertically. Symptoms of acute HBV infection include slight fever (which may be absent), malaise, and mild gastrointestinal upset. Visible jaundice is usually the first significant finding and is accompanied by darkening of the urine and pale or clay-colored stools. Hepatomegaly is frequently present. Occasionally an antigen-antibody complex presentation antedates the appearance of icterus, and is characterized by macular rash, urticaria, and arthritis. HBV infection more rarely presents as a glomerulonephritis or nephrotic syndrome from immune complexes.
B. Laboratory Findings
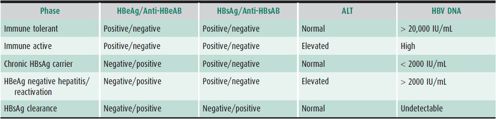
The diagnosis of acute HBV infection is confirmed by the presence of HBsAg and anti-HBc IgM. Recovery from acute infection is accompanied by HBsAg clearance and appearance of anti-HBs and anti-HBc IgG. Individuals who are immune by vaccination are positive for anti-HBs, but negative for anti-HBc IgG. Chronic infection is defined as the presence of HBsAg for at least 6 months. Vertical transmission to newborns is documented by positive HBsAg. LFT results are similar to those discussed earlier for hepatitis A. Liver biopsy is most useful in chronic infection to determine the degree of fibrosis and inflammation. Renal involvement may be suspected on the basis of urinary findings suggesting glomerulonephritis or nephrotic syndrome. The various phases of chronic HBV infection are shown in Table 22–7.
Table 22–7. Phases of chronic hepatitis B infection.
 Differential Diagnosis
Differential Diagnosis
The differentiation between HAV and HBV disease is aided by a history of parenteral exposure, an HBsAg-positive parent, or an unusually long period of incubation. HBV and hepatitis C virus (HCV) infection or Epstein-Barr virus (EBV) infection are differentiated serologically. The history may suggest a drug-induced hepatitis, especially if a serum sickness prodrome is reported. Autoimmune hepatitis Wilson disease, hemochromatosis, nonalcoholic fatty liver disease (NAFLD), α1-antitrypsin deficiency, and drug-induced hepatitis should also be considered.
 Treatment
Treatment
Supportive measures such as bed rest and a nutritious diet are used during the active symptomatic stage of acute HBV infection. Corticosteroids are contraindicated. No other treatment is needed for acute HBV infection. For acute infection complicated by acute liver failure, nucleos(t)ide therapy may be helpful. For patients with chronic infection who persist in the immunoactive phase for more than 6 months or with HBeAg-negative chronic hepatitis, there are currently two approved treatment options. Treatment with α-interferon (5–6 million U/m2 of body surface area injected subcutaneously three times a week for 4–6 months) inhibits viral replication in 30%–40% of patients, normalizes the ALT level, and leads to the disappearance of HBeAg and the appearance of anti-HBe. Side effects are common. Younger children may respond better than older children. Orally administered nucleoside analog therapy (lamivudine 3 mg/kg/d up to 100 mg/d for children > 2 years old or adefovir 10 mg/d or tenofovir 300 mg/d for children > 12 years old and entecavir [0.5 mg once daily] or telbivudine [600 mg once daily] for children > 16 years old) leads to a successful response in 25% of treated children, with minimal side effects, but may require long-term treatment. However, resistant organisms can emerge, more frequently with lamivudine. Pegylated interferon, several oral antiviral agents (with much lower rates of resistance), and combination therapy are promising options being tested in children. Immunotolerant children and chronic carriers do not respond to therapy. Liver transplantation is successful in acute liver failure due to hepatitis B; however, reinfection is common following liver transplantation for chronic hepatitis B unless long-term HBIG or antivirals are used.
 Prognosis
Prognosis
The prognosis for acute HBV infection is good in older children, although acute liver failure (< 0.1%) or chronic hepatitis and cirrhosis (up to 10%) may supervene. The course of the acute disease is variable, but jaundice seldom persists for more than 2 weeks. HBsAg disappears in 95% of cases at the time of clinical recovery. Chronic infection is particularly common in children with vertical transmission, Down syndrome, or leukemia, and in those undergoing chronic hemodialysis. Persistence of neonatally acquired HBsAg occurs in 70%–90% of infants without immunoprophylaxis or vaccination. The presence of HBeAg in the HBsAg carrier indicates ongoing viral replication. However, 1%–2% of children infected at birth will show spontaneous seroconversion of HBeAg each year. If HBV infection is acquired later in childhood, HBV is cleared and recovery occurs in 90%–95% of patients. Chronic HBV disease predisposes the patient to development of hepatocellular carcinoma. Once chronic HBV infection is established, surveillance for development of hepatocellular carcinoma with serum α-fetoprotein is performed biannually and ultrasonography yearly. Routine HBV vaccination of newborns in endemic countries has reduced the incidence of acute liver failure, chronic hepatitis, and hepatocellular carcinoma in children.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


