0
No contraction
1
Flicker or trace movement
2
Active movement with gravity eliminated
3
Active movement against gravity
4
Active movement against gravity and resistance
5
Normal power
The MRC is useful in children that are able to cooperate with an examination and provide reliable effort. Thus this test is more helpful in children over 4 or 5 years of age. The scale above is also adapted by providers where (−) and (+) signs are used to further subdivide strength within a given score in the 2–5 range.
While this chapter will not delve into all upper extremity rating scales or functional rating scales (see chapter “Occupational Therapy Evaluation and Treatment”), it is useful to note a “functional” rating scale that describes upper extremity function that has been developed for the evaluation of boys with Duchenne muscular dystrophy. The Brooke scale was designed to assess upper extremity function with a score from 1 (least severe) to 6 (most severe). This scale was designed for progression of upper extremity dysfunction in Duchenne muscular dystrophy (Brooke et al. 1981). While it could theoretically be applied to some of the other inherited neuromuscular conditions, caution would need to be used, as other disorders may have early contractures that limit function thus rendering a far lower score earlier in disease course. See Table 2.
Table 2
Brooke scale of upper extremity function
Grade | Description |
|---|---|
1 | Starting with arms at the sides, the patient can abduct the arms in a full circle until they touch above the head |
2 | Can raise arms above head only by flexing the elbow (shortening the circumference of the movement) or using accessory muscles |
3 | Cannot raise hands above head, but can raise an 8-oz glass of water to the mouth |
4 | Can raise hands to the mouth, but cannot raise an 8-oz glass of water to the mouth |
5 | Cannot raise hands to the mouth, but can use hands to hold a pen or pick up pennies from the table |
6 | Cannot raise hands to the mouth and has no useful function of hands |
In addition to evaluating strength and tone, it is also important to take note of muscle bulk. Patients may have certain patterns of muscle atrophy or pseudohypertrophy that may be characteristic of particular types of diseases. Further details on the history and examination of patients with neuromuscular disease will be provided later in this chapter.
Diagnoses
This chapter is focusing on those disorders of muscle and so will not address disorders of the motor neuron, nerves, or neuromuscular junction that can all affect upper extremity function. For the purpose of clarity and focus, this chapter will not cover mitochondrial disorders and will not cover all metabolic disorders that can affect muscle, but will focus on one (Pompe disease). The inherited disorders of muscle that will be discussed in this chapter will include congenital muscular dystrophies, congenital myopathies, myotonic dystrophy, dystrophinopathies, Emery-Dreifuss muscular dystrophy, and limb-girdle muscular dystrophies. In general, with the exception of myotonic dystrophy and Pompe disease, most of the disorders discussed in this chapter are caused by abnormalities in the contractile apparatus, nuclear membrane, connection between the contractile apparatus and the sarcolemmal membrane, membrane repair, and extracellular matrix. See Fig. 1 which shows a diagram of the proteins contributing to various inherited muscle diseases. The disorders discussed in this chapter will largely be pediatric-onset disorders.
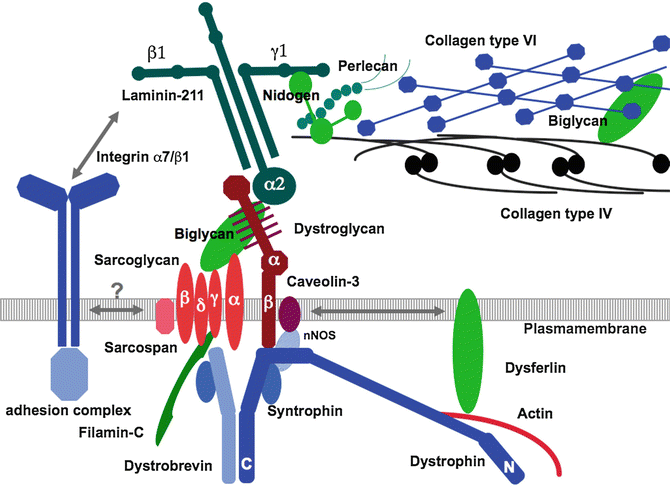
Fig. 1
Diagram of the proteins near the Sarcolemnal (Plasma) membrane (Seen ingray). Note, extracellular proteins are seen above and intracellular proteins are seen below. The represent some (but not all) proteins that contribute to various forms of inherited muscle disease.
Congenital Onset
The congenital myopathies and congenital muscular dystrophies are defined based upon clinically apparent muscle weakness at birth or in early infancy. While some patients may not be recognized to be symptomatic until a later age, the onset of disease is the one that begins prenatally or in infancy. There can be varying degrees of severity from very severe prenatal onset to subtle motor difficulties.
The congenital myopathies are a group of disorders often with characteristic histopathological findings on muscle biopsy and typically marked by a disease course that is relatively stable over longer periods of follow-up. While historically these disorders have been defined by the characteristic histological findings, it is important to note that the absence of such findings does not always preclude some of these diagnoses. There can be a range of histopathological features seen in these disorders, and the variability in histological findings within particular disorders may partly be explained by the age of biopsy and the site of biopsy. Symptoms can often begin prenatally, with findings of polyhydramnios, decreased fetal movement, and breech position. At birth, findings of arthrogryposis of the upper and lower extremity may be seen, as well as hip dislocations and osteopenia related to poor fetal movement, sometimes leading to fractures under delivery. Within this group of disorders, there is great clinical variability, with many of the entities ranging from a severe and even lethal neonatal form with fetal akinesia to a later onset or milder form. At the most severe end of the spectrum (of those who survive in utero to delivery) are infants who have trace to no movements of the extremities, severe inability to feed, and respiratory failure. Patients may have ophthalmoplegia, in particular in the centronuclear myopathies (Pierson et al. 2005). Some patients can have pronounced facial weakness as well. CK levels are often normal or can be mildly elevated. See Table 3 for a list of genetic causes and “classic” histological features seen in the congenital myopathies (Pierson et al. 2005; Jungbluth et al. 2003; Goebel 2005). Overall, patients with congenital myopathy tend to be stable over long periods of time and even make motor gains. The predominant factors that correlate with mortality or morbidity in this group of disorders are the existence of severe respiratory failure and risk of aspiration (to food or secretions).
Table 3
Congenital myopathies
Disease | Gene | Salient clinical features | Classical histological findings |
|---|---|---|---|
Nemaline myopathy | ACTA1, TPM3, NEB, TPM2, TNNT1, CFL2, KBTBD13, KBTBD5 | Range in severity | Nemaline rods – in continuity with Z lines |
Classic form facial and axial muscle weakness with onset in infancy or childhood | |||
Feeding difficulties common | |||
Severe respiratory involvement may be seen | |||
Central core disease/RYR1 associated myopathy | RYR1 | Variable severity (severe neonatal- to childhood-onset hypotonia and delayed motor milestones) | Classic “cores” – areas of clearing on oxidative stains (NADH) |
Possible ophthalmoplegia | May also see multi-minicores | ||
Severe respiratory involvement may be seen | Absence of cores does not preclude diagnosis | ||
Risk of malignant hyperthermia | |||
Sparing of rectus femoris muscle on muscle imaging | |||
Centronuclear myopathy/myotubular myopathy | MTM1 | X-linked (severely affected males) | Centrally placed nuclei |
Severe neonatal hypotonia | |||
Ophthalmoplegia | |||
Early respiratory failure/ventilator dependence | |||
Centronuclear myopathy | BIN1, DNM2, RYR1 | Severe neonatal hypotonia (variable) | Centrally placed nuclei |
Congenital fiber-type disproportion | TPM3 | Hypotonia at birth or shortly after | Type 1 fiber hypotrophy (12 % smaller than the type 2 fibers |
ACTA1 | Variable degree of weakness | ||
SEPN1 | Static or may show improvement |
The congenital muscular dystrophies are a heterogeneous group of disorders where the muscle reveals dystrophic or myopathic features without clear structural changes consistent with congenital myopathies. The main congenital muscular dystrophies include the following disorders: disorders of alpha-dystroglycan glycosylation (which can have significant brain involvement), collagen VI disorders, and laminin α2 CMD. See Table 4 for a list and summary of these disorders, key clinical features, and genetic causes. Collagen VI-related congenital muscular dystrophy has more striking upper extremity involvement (involving contractures and joint laxity) that will be further discussed in section “Diagnostic Features in Inherited Muscle Disease and Concepts of Upper Extremity Involvement” of this chapter.
Table 4
Congenital muscular dystrophies
Disease | Gene symbol [protein product] | Motor features | Ocular features | Salient neurological features | MRI brain findings (if applicable) |
|---|---|---|---|---|---|
α-Dystroglycanopathies | |||||
Walker-Warburg syndrome (WWS) | POMT1 [protein O-mannosyltransferase 1] | Severe hypotonia | Poor visual attention | Decreased alertness | Agyria or severe lissencephaly |
POMT2 [protein O-mannosyltransferase 2] | Neonatal onset | Retinal dysgenesis | Severe intellectual disability | Polymicrogyria | |
FKTN [fukutin] | Absent psychomotor acquisition | Microphthalmia | White matter – dysmyelination or cystic changes | ||
LARGE [acetylglucosaminyltransferase-like protein] | Congenital cataracts | Cerebellar/brainstem hypoplasia | |||
Hydrocephalus | |||||
Complete or partial absent corpus callosum | |||||
Muscle-eye-brain disease (MEB)/Fukuyama CMD (FCMD) | POMGnT1 | Hypotonia at birth or 1st few months | Milder than WWS | Severe intellectual disability | Variable (mild to severe changes) |
POMT1 | Ambulation may acquired | Congenital glaucoma | Refractory epilepsy | Frontoparietal pachygyria | |
POMT2 | Progressive myopia | Behavioral problems | Polymicrogyria | ||
ISPD [isoprenoid synthase domain-containing protein | Retinal atrophy | Joint contractures | Cerebellar/brainstem hypoplasia | ||
FKRP | Juvenile cataracts | Cerebellar vermis hypoplasia/dysplasia/cysts | |||
FKTN | Rarely severe | Periventricular white matter changes | |||
CMD-MR (CMD with mental retardation) | POMT1 | Neonatal-onset hypotonia | Ocular findings rare/mild | Mild cognitive impairment | Isolated microcephaly |
Minor white matter changes | |||||
CMD-no MR | Fukutin | Neonatal- or childhood-onset hypotonia or delayed motor development | Normal or mild | Normal or minimal impairment | Normal or mild abnormality |
FKRP | |||||
Defects of extracellular matrix proteins | |||||
LAMA2-related CMD (MDC1A, merosin-deficient CMD) | LAMA2 | Neonatal-onset max motor ability – sit and stand with support | N/A | Can have neuropathy, epilepsy, subclinical cardiomyopathy | Abnormal white matter signal |
[Laminin α2] | Occipital pachygyria | ||||
Pontocerebellar atrophy (rare) | |||||
COL6-related dystrophy (Ullrich CMD and Bethlem myopathy) | COL6A1, COL6A2, COL6A3 | Variable severity (ambulatory to difficulty sitting and severe respiratory failure) | N/A | Distal joint laxity | None |
[Collagen VI] | Proximal contractures | ||||
Keratosis pilaris | |||||
Defects of proteins of the endoplasmic reticulum | |||||
Rigid spine muscular dystrophy | SEPN1 | Delayed walking | N/A | Rigid spine | None |
[Selenoprotein] | Axial weakness | Restrictive lung disease | |||
Slow progression | |||||
Defects of nuclear envelope proteins | |||||
Lamin A/C-related CMD | LMNA | Axial weakness | N/A | Early motor deterioration | None |
[Lamin A/C] | “Dropped head” syndrome – posterior neck weakness | Spinal rigidity | |||
In general this group of disorders tends to be more progressive in nature. The onset is typically either prenatally, at birth, or within the first year of life. Clinical features include diffuse weakness, contractures (either in infancy or some that develop later in childhood), and potential central nervous system involvement in some (e.g., developmental delays, seizures, spasticity). The maximum motor skill achieved can vary. In the more severe of the congenital disorders of muscle, ambulation may not be achieved, while in a more moderate group ambulation may be achieved and later lost. In those representing the severe end of the spectrum, patients may also have significant upper extremity involvement – and yet the patients are significantly dependent on upper extremity function for tasks such as feeding, use of wheelchair, writing, typing, and other activities of daily living. Children may make motor gains early in life, but will eventually show some decline in function. The rate of decline and age of decline vary by disease and also from patient to patient within the same disease. The degree of deterioration and degree of impairment also vary greatly in this group of disorders. Pathophysiologically, subgroups of disorders in this category are caused by abnormalities of the extracellular matrix of the muscle cell membrane, intracellular molecules, and inner nuclear matrix proteins. See Table 4 which will review the genetic causes of congenital muscular dystrophy (Sparks et al. 2012; Godfrey et al. 2007). Several of the alpha-dystroglycanopathies represent the more severe end of the spectrum with neonatal onset of severe weakness, cognitive impairment, visual impairment, and CNS MRI changes. CMDs with more striking and unique upper extremity involvement include the collagen VI disorders where there is distal hyperlaxity combined with proximal upper extremity contractures that are progressive in nature. This will be discussed further in a later section of this chapter.
In addition to the abovementioned disorders, myotonic dystrophy is a disorder that can have congenital onset or later in life onset (as there is anticipation and increased severity from one generation to the next). Myotonic dystrophy type 1 (DM1) and type 2 (DM2) are autosomal dominant multisystem disorders. DM1 is caused by expansion of a CTG trinucleotide repeat in the noncoding region of the DMPK gene, with increased repeats correlating to disease severity. DM1 can affect skeletal muscle, smooth muscle, cardiac muscle, as well as the eye, endocrine system, and central nervous system (Bird 2013). Clinical findings and severity in DM1 are variable from mild to severe, with three predominant phenotypes: mild, classic, and severe congenital. In classic DM1, weakness and muscle wasting of facial muscles (especially temporal wasting), atrophy of the forearm flexor muscles, weakness of long finger flexors, and weakness of ankle dorsiflexors can be seen. Patients can have prominent myotonia, but this is often not present on examination of neonates with suspected congenital myotonic dystrophy. In its most severe form, in those patients with congenital myotonic dystrophy, severe neonatal hypotonia and respiratory failure with feeding difficulties can be seen. In patients with congenital DM1, weakness is more diffuse, but most notably, there is very striking facial weakness as compared to the typical or later onset DM1. Respiratory failure and prolonged ventilator dependence in patients with congenital DM1 correlates with higher rates of mortality. Patients with myotonic dystrophy, especially those with the congenital form, often have intellectual disability, personality disorder, cataracts, and cardiac conduction deficits. DM2 tends to be milder and have a later onset in life. DM2 is characterized by myotonia and muscle dysfunction, with onset in childhood or adult years, and is less commonly associated with cardiac conduction defects, cataracts, or insulin insensitivity (as is seen in DM1). The onset of myotonia tends to be later in life, often not until the third decade. Muscle weakness and pain may be episodic or fluctuating, with predominant involvement of neck flexors, finger flexors, elbow extensors, and hip flexors and extensors (Dalton et al. 2013). Expansion of the CCTG repeat on the CNBP (ZNF9) gene causes DM2.
Childhood Onset
In contrast to those disorders with onset perinatally or in infancy, there are certain muscle diseases that more distinctly have onset during the childhood, adolescent, or adult years. This includes dystrophinopathies, myotonic dystrophy (although this can also have a severe congenital form as discussed above), Emery-Dreifuss muscular dystrophy, FSHD, Pompe disease, and limb-girdle muscular dystrophies (LGMDs). While there are some exceptions, these are a group of disorders predominantly affecting the shoulder and pelvic girdle muscles with age of onset in childhood, adolescence, or even adulthood. This group of disorders is marked by gradual progression of weakness over time. Although proximal muscles are affected first, over time, distal muscles can become involved. CK levels generally tend to be elevated. Muscle biopsy reveals dystrophic changes, and immunohistochemical stains can be helpful in narrowing down the diagnosis.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


