13.2 Immunodeficiency and its investigation
Host factors and resistance to infection
1. Innate, non-antigen-specific responses are initiated early. There are an increasing number of recognized components, including:
2. Adaptive, antigen-specific immune responses are the basis of immunological memory and are essential for maturation of protective immune responses and efficacy of vaccination. The components are:
Deficiencies or disruption of any of these components can predispose to infection. These defects may be the result of immaturity, primary or acquired deficiency, influencing age and severity of presentation as well as management and prognosis. Some disorders will result in localized disease, whereas others predispose to infection with specific microorganisms, as shown in Figure 13.2.1.
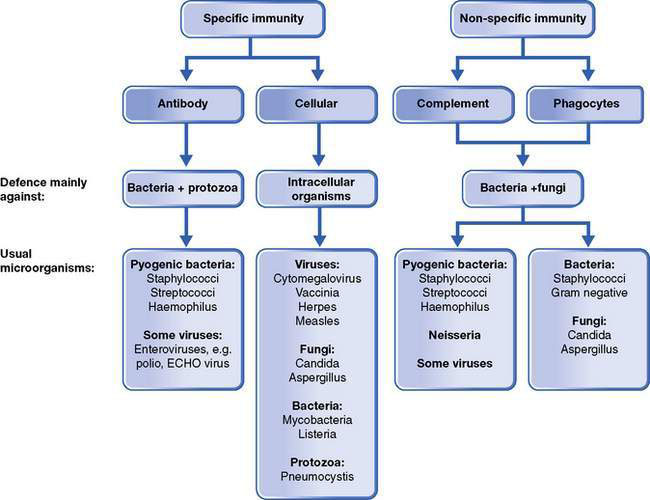
Age of presentation, infective complications and diagnosis
Deficiencies of humoral immunity typically present after the age of 4–5 months, when maternally-derived antibody has waned. Significant deficiencies of T-cell function present earlier, within the first months. The type of infection(s) as well as associated clinical features may provide clues to the deficient immune component (see Fig. 13.2.1, Table 13.2.1). In children presenting with recurrent fever without obvious and/or identifiable infection, the possibility of an autoinflammatory syndrome should be considered. Many primary immunodeficiencies present in infancy with dermatological manifestations such as severe or atypical eczema, thrombocytopenic purpura, recalcitrant candidiasis and abscesses. Some are diagnosed in association with other conditions such as cardiac, endocrine and neurological anomalies (e.g. DiGeorge syndrome, ataxia telangiectasia). Conditions such as common variable immunodeficiency, natural killer cell and complement deficiencies can present at a range of ages, from late infancy to young adulthood.
Table 13.2.1 Clinical features suggestive of some immune defects
| Clinical feature | Immunodeficiency |
|---|---|
| Recurrent sinopulmonary infections/chronic diarrhoea and failure to thrive (and, less commonly, cytopenias, arthritis, hepatitis, coeliac disease, inflammatory bowel disease, granuloma formation, malignancy) | Humoral |
| Recurrent fungal, opportunistic infections/chronic diarrhoea, failure to thrive, neonatal hypocalcaemia | Cellular |
| Recurrent periodontal disease, gingivitis, skin and deep abscesses, fungal pneumonia, osteomyelitis | Phagocytic |
| Recurrent or severe meningococcal or pneumococcal infection | Complement |
Defects associated with prematurity and delays in immunological development
Physiological hypogammaglobulinaemia of infancy occurs between 3 and 6 months of life at the nadir of waning maternal IgG balanced by increasing infant production of IgG (Fig. 13.2.2). This can be accentuated and prolonged in premature infants by a reduced store of maternally derived IgG, or alternatively by delay in IgG production, the latter termed transient hypogammaglobulinaemia of infancy.
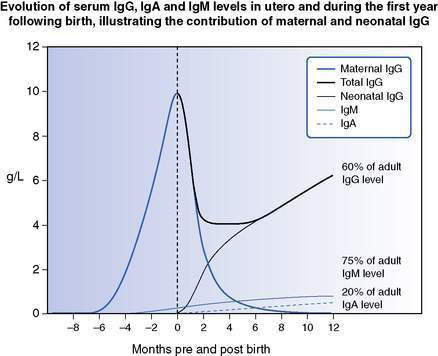

Primary and secondary immunodeficiencies
More than 150 primary immunodeficiencies (PID) have been identified and characterized, and the number is growing constantly. An expert international committee of the International Union of Immunological Societies (IUIS) meets regularly to update continually the known primary immunodeficiencies and, where identified, the underlying genetic cause. The publication by Notarangelo and co-workers, summarizing the most recent meeting in 2009, contains detailed tables summarizing clinical features, laboratory findings, genetics and relative frequency of known PIDs. Although, individually, most are rare or extremely rare, many affect similar pathways or categories of immune function and thus collectively are not uncommon. Table 13.2.2 lists these conditions as categorized by IUIS and, where known, the mode of inheritance.
Table 13.2.2 International Union of Immunological Societies (IUIS) classification of primary immunodeficiencies
| Disease | Inheritance |
|---|---|
| Combined T- and B-cell immunodeficiency | |
| T− B+ SCID | |
| γc deficiency | XL |
| JAK3 deficiency | AR |
| IL-7 receptor α deficiency | AR |
| CD45 deficiency | AR |
| CD3δ/CD3ε/CD3ζ deficiency | AR |
| Coronin-1A deficiency | AR |
| T− B− SCID | |
| RAG1/RAG2 deficiency | AR |
| DCLREIC (Artemis) deficiency | AR |
| DNA PKcs deficiency | AR |
| Adenosine deaminase (ADA) deficiency | AR |
| Reticular dysgenesis | AR |
| Omenn syndrome | AR (most) |
| DNA ligase IV deficiency | AR |
| Cernunnos deficiency | AR |
| CD40 ligand deficiency | XL |
| CD40 deficiency | AR |
| Purine nucleoside phosphorylase deficiency | AR |
| CD3γ deficiency | AR |
| CD8 deficiency | AR |
| ZAP-70 deficiency | AR |
| Ca2+ channel deficiency | AR |
| MHC class I deficiency | AR |
| MHC class II deficiency | AR |
| Winged helix deficiency (nude) | AR |
| CD25 deficiency | AR |
| STAT5b deficiency | AR |
| Itk deficiency | AR |
| DOCK8 deficiency | AR |
| Predominantly antibody deficiencies | |
| Severe reduction in all serum Ig isotypes with profoundly decreased or absent B cells | |
| Btk deficiency | XL |
| μ heavy chain deficiency | AR |
| λ5 deficiency | AR |
| Igα deficiency | AR |
| Igβ deficiency | AR |
| BLNK deficiency | AR |
| Thymoma with immunodeficiency | None |
| Severe reduction in at least 2 serum Ig isotypes with normal or low numbers of B cells | |
| Common variable immunodeficiency disorders | Variable |
| ICOS deficiency | AR |
| CD19 deficiency | AR |
| TACI deficiency | AD or AR |
| BAFF receptor deficiency | AR |
| Severe reduction in serum IgG and IgA with normal/raised IgM and normal B-cell numbers | |
| CD40L deficiency | XL |
| CD40 deficiency | AR |
| AID deficiency | AR |
| UNG deficiency | AR |
| Isotype or light chain deficiencies with normal numbers of B cells | |
| Ig heavy chain mutations or deletions | AR |
| κ chain deficiency | AR |
| Isolated IgG subclass deficiency | Variable |
| IgA with IgG subclass deficiency | Variable |
| Selective IgA deficiency | Variable |
| Specific antibody deficiency with normal Ig concentrations and normal B cell numbers | Variable |
| Transient hypogammaglobulinaemia of infancy with normal B-cell numbers | Variable |
| Other well defined immunodeficiency syndromes | |
| Wiskott–Aldrich syndrome | XL |
| DNA repair defects | |
| Ataxia telangectasia | AR |
| Ataxia telangectasia-like disease | AR |
| Nijmegen breakage syndrome | AR |
| Bloom syndrome | AR |
| Immunodeficiency with centromeric instability and facial anomalies (ICF) | AR |
| PMS2 deficiency | AR |
| Thymic defects | |
| DiGeorge anomaly | De novo/AD |
| Immune osseous dysplasias | |
| Cartilage hair hypoplasia | AR |
| Schimke syndrome | AR |
| Comel–Netherton syndrome | AR |
| Hyper-IgE syndromes (HIES) | |
| AD-HIES | AD |
| AR-HIES | AR |
| Chronic mucocutaneous candidiasis | AD/AR/sporadic |
| Hepatic veno-occlusive disease with immunodeficiency | AR |
| XL dyskeratosis congenita | XL |
| Diseases of immune dysregulation | |
| Immunodeficiency with hypopigmentation | |
| Chediak–Higashi syndrome | AR |
| Griscelli syndrome, type 2 | AR |
| Hermansky–Pudlak syndrome, type 2 | AR |
| Familial haemophagocytic lymphohistiocytosis | |
| Perforin deficiency | AR |
| UNC13-D deficiency | AR |
| Syntaxin 11 deficiency | AR |
| Lymphoproliferative syndromes | |
| XLP1, SH2D1A deficiency | XL |
| XLP2, XIAP deficiency | XL |
| Itk deficiency | AR |
| Syndromes with autoimmunity | |
| Autoimmune lymphoproliferative syndrome (ALPS) | |
| CD95 (Fas) defects, ALPS type 1a | AD |
| CD95L (Fas ligand) defects, ALPS type 1b | AR |
| Caspase 10 deficiency, ALPS type 2a | AR |
| Caspase 8 deficiency, ALPS type 2b | AR |
| Activating N-ras, N-ras-dependent ALPS | AD |
| APECED (autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy) | AR |
| IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) | XL |
| CD25 deficiency | AR |
| Congenital defects of phagocyte number, function or both | |
| Severe congenital neutropenias | AD |
| Kostman disease | AR |
| Neutropenia with cardiac and urogenital malformations | AR |
| Glycogen storage disease type 1b | AR |
| Cyclic neutropenia | AR |
| X-linked neutropenia/myelodysplasia | AR |
| P14 deficiency | AR |
| Leukocyte adhesion deficiency type 1 | AR |
| Leukocyte adhesion deficiency type 2 | AR |
| Leukocyte adhesion deficiency type 3 | AR |
| Rac2 deficiency | AR |
| β-Actin deficiency | AR |
| Localized juvenile periodontitis | AR |
| Papillon–Lefèvre syndrome | AR |
| Specific granule deficiency | AR |
| Schwachman–Diamond syndrome | AR |
| X-linked chronic granulomatous disease | AR |
| Autosomal chronic granulomatous diseases | AR |
| IL-12 and IL-23 receptor β1-chain deficiency | AR |
| IL-12p40 deficiency | AR |
| IFN-γ receptor 1 deficiency | AR, AD |
| IFN-γ receptor 2 deficiency | AR |
| STAT1 deficiency | AR |
| AR hyper-IgE syndrome | AR |
| AD hyper-IgE syndrome | AD |
| Pulmonary alveolar proteinosis | Bi-allelic |
| Defects in innate immunity | |
| Anhidrotic ectodermal dysplasia with immunodeficiency (EDA-ID) | XL |
| AD EDA-ID | AD |
| IL-1 receptor-associated kinase 4 (IRAK4) deficiency | AR |
| MyD88 deficiency | AR |
| WHIM (warts, hypogammaglobulinaemia, infections, myelokathexis syndrome) | AD |
| Epidermodysplasia verruciformis | AR |
| Herpes simplex encephalitis (HSE) – UNC93B1 | AR |
| HSE – TLR3 | AD |
| Chronic mucocutaneous candidiasis – CARD9 | AR |
| Trypanosomiasis | AD |
| Autoinflammatory disorders | |
| Familial Mediterranean fever | AR |
| TNF receptor-associated periodic fever syndrome (TRAPS) | AD |
| Hyper-IgD syndrome | AR |
| Muckle–Wells syndrome | AD |
| Familial cold autoinflammatory syndrome | AD |
| NOMID/CINCA | AD |
| Pyogenic sterile arthritis, pyoderma gangrenosum, acne (PAPA) syndrome | AD |
| Blau syndrome | AD |
| Chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed) | AR |
| DIRA (deficiency of IL-1 receptor antagonist) | AR |
| Complement deficiencies | |
| C1q deficiency | AR |
| C1r deficiency | AR |
| C1s deficiency | AR |
| C4 deficiency | AR |
| C2 deficiency | AR |
| C3 deficiency | AR |
| C5 deficiency | AR |
| C6 deficiency | AR |
| C7 deficiency | AR |
| C8 deficiency | AR |
| C9 deficiency | AR |
| C1 inhibitor deficiency | AR |
| Factor I deficiency | AR |
| Factor H deficiency | AR |
| Factor D deficiency | AR |
| Properdin deficiency | AR |
| Mannose-binding lectin deficiency | AR |
| MASP2 deficiency | AR |
| Complement receptor 3 (CR3) deficiency | AR |
| Membrane cofactor protein (CD46) deficiency | AR |
| Membrane attack complex inhibitor (CD59) deficiency | AR |
| Paroxysmal nocturnal haemoglobinuria | Acquired |
| Immunodeficiency associated with ficolin-3 deficiency | AR |
AD, autosomal dominant; AR, autosomal recessive; IFN, interferon; Ig, immunoglobulin; IL, interleukin; XL, X-linked.
Modified from: Notarangelo LD, Fischer A, Geha RS et al 2009 Primary immunodeficiencies: 2009 update. Journal of Allergy and Clinical Immunology 124:1161–1178.
An underlying PID may be suggested by the frequency, severity and type of infection, the response or lack of response to antimicrobial therapy, associated failure to thrive and existence of significant family history (Box 13.2.1).
Box 13.2.1 Warning signs of primary immunodeficiency
1. Eight or more new ear infections within 1 year
2. Two or more serious sinus infections within 1 year
3. Two or more months on antibiotics with little effect
4. Two or more pneumonias within 1 year
5. Failure of an infant to gain weight or grow normally
6. Recurrent, deep skin or organ abscesses
7. Persistent thrush in the mouth or on the skin, after age 1 year
8. Need for intravenous antibiotics to clear infections
9. Two or more deep-seated infections such as meningitis, osteomyelitis, cellulitis or sepsis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


