Chapter 551 Hypopituitarism
Hypopituitarism denotes underproduction of growth hormone (GH) alone or in combination with deficiencies of other pituitary hormones. Affected children have postnatal growth impairment that is specifically corrected by replacement of GH. The incidence of congenital hypopituitarism is thought to be between 1 in 4,000 and 1 in 10,000 live births. With expanding knowledge of the genes that direct pituitary development or hormone production, an increasing proportion of cases can be attributed to specific genetic disorders. Mutations in 7 candidate genes account for 13% of isolated growth hormone deficiency (IGHD) and 20% of multiple pituitary hormone deficiency (MPHD) cases. The likelihood of finding mutations is increased by positive family histories and decreased in cases with adrenocorticotropin hormone (ACTH) deficiency. The genes, hormonal phenotypes, associated abnormalities and modes of transmission for such established genetic disorders are shown in Tables 551-1 and 551-2. Acquired hypopituitarism usually has a later onset and different causes (Table 551-3).
Table 551-1 ETIOLOGIC CLASSIFICATION OF MULTIPLE PITUITARY HORMONE DEFICIENCY
| GENE OR LOCATION | PHENOTYPE | INHERITANCE |
|---|---|---|
| GENETIC FORMS | ||
| POU1F1 (PIT1) | GH, TSH, PRL | R, D |
| PROP1 | GH, TSH, PRL, LH, FSH, ±ACTH, variable AP | R |
| LHX3 | GH, TSH, PRL, LH, FSH, variable AP, ±short neck | R |
| LHX4 | GH, TSH, ACTH, small AP, EPP, ±Arnold Chiari | D |
| TPIT | ACTH, severe neonatal form | R |
| HESX1 | GH, variable for others, small AP, EPP | R, D |
| SOX3 | Variable deficiencies, ±MR, EPP, small AP and stalk | XL |
| PTX2 | Rieger syndrome | D |
| GLI2 | Holoprosencephaly, midline defects | D |
| GLI3 | Hall-Pallister syndrome | D |
| SHH (Sonic hedgehog) | GH deficiency with single central incisor | D |
| ACQUIRED FORMS | ||
| Idiopathic | ||
| Irradiation | GH deficiency precedes other deficiencies | |
| Inflammation | Histiocytosis, sarcoidosis | |
| Autoimmune | Hypophysitis | |
| Post-surgical | Stalk section, vascular compromise | |
| Tumor | Craniopharyngioma, glioma, pinealoma | |
| Trauma | Battering, shaken baby, vehicular | |
| UNCERTAIN ETIOLOGY | ||
| Idiopathic | ||
| Congenital absence of pituitary | ||
| Septo-optic dysplasia | ||
| Birth trauma | ||
ACTH, adrenocorticotropic hormine; AP, anterior pituitary; D, dominant; EPP, ectopic posterior pituitary; FSH, follicle-stimulating hormone; GH, growth hormone; LH, luteinizing hormone; MR, mental retardation; PRL, prolactin; R, recessive; TSH, thyroid-stimulating hormone; XL, X-linked.
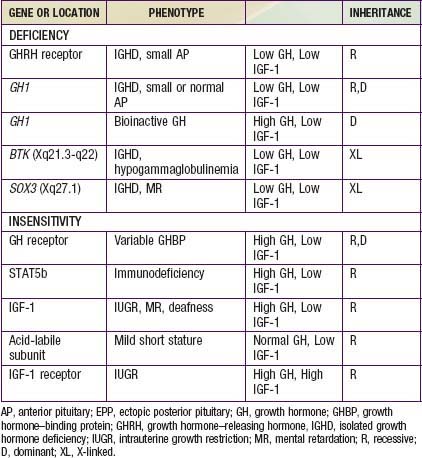
Table 551-3 CAUSES OF ACQUIRED HYPOPITUITARISM
BRAIN DAMAGE*
PITUITARY TUMORS*
NON-PITUITARY TUMORS
INFECTION
INFARCTION
AUTOIMMUNE DISORDER
OTHER
* Pituitary tumors are classically the most common cause of hypopituitarism. However, new findings imply that causes related to brain damage might outnumber pituitary adenomas in causing hypopituitarism.
From Schneider HJ, Aimaretti G, Kreitschmann-Andermahr I, et al: Hypopituitarism, Lancet 369:1461–1470, 2007.
Multiple Pituitary Hormone Deficiency
Genetic Forms
HESX1
The HESX1 gene is expressed in precursors of all 5 cell types of the anterior pituitary early in embryologic development. Mutations result in a complex phenotype with defects in development of the optic nerve. Heterozygotes for loss-of-function mutations show the combinations of isolated GH deficiency and optic nerve hypoplasia. Homozygotes can have full expression of septo-optic dysplasia (SOD). This condition combines incomplete development of the septum pellucidum with optic nerve hypoplasia and other midline abnormalities. Clinical observation of nystagmus and visual impairment in infancy leads to the discovery of optic nerve and brain abnormalities. SOD is associated with anterior and/or posterior pituitary hormone deficiencies in about 25% of the cases. These patients often show the triad of a small anterior pituitary gland, an attenuated pituitary stalk, and an ectopic posterior pituitary bright spot. The great majority of patients with SOD do not have HESX1 mutations. The etiology might involve mutations in another gene or a nongenetic explanation (Chapters 585 and 623).
Other Congenital Forms
Pituitary hypoplasia can occur as an isolated phenomenon or in association with more extensive developmental abnormalities such as anencephaly or holoprosencephaly. Midfacial anomalies (cleft lip, palate; Chapter 302) or the finding of a solitary maxillary central incisor indicate a high likelihood of GH or other anterior or posterior hormone deficiency. At least 12 genes have been implicated in the complex genetic etiology of holoprosencephaly (Chapter 585.7). In the Hall-Pallister syndrome, absence of the pituitary gland is associated with hypothalamic hamartoblastoma, postaxial polydactyly, nail dysplasia, bifid epiglottis, imperforate anus, and anomalies of the heart, lungs, and kidneys. The combination of anophthalmia and hypopituitarism has been associated with mutations in the SIX6, SOX2, and OTX2 genes.
Acquired Forms
Any lesion that damages the hypothalamus, pituitary stalk, or anterior pituitary can cause pituitary hormone deficiency (see Table 551-3). Because such lesions are not selective, multiple hormonal deficiencies are usually observed. The most common lesion is the craniopharyngioma (Chapter 491). Central nervous system germinoma, eosinophilic granuloma (histiocytosis), tuberculosis, sarcoidosis, toxoplasmosis, meningitis, and aneurysms can also cause hypothalamic-hypophyseal destruction. Trauma, including shaken child syndrome (Chapter 37), motor vehicle accidents, traction at delivery, anoxia, and hemorrhagic infarction, can also damage the pituitary, its stalk, or the hypothalamus.



