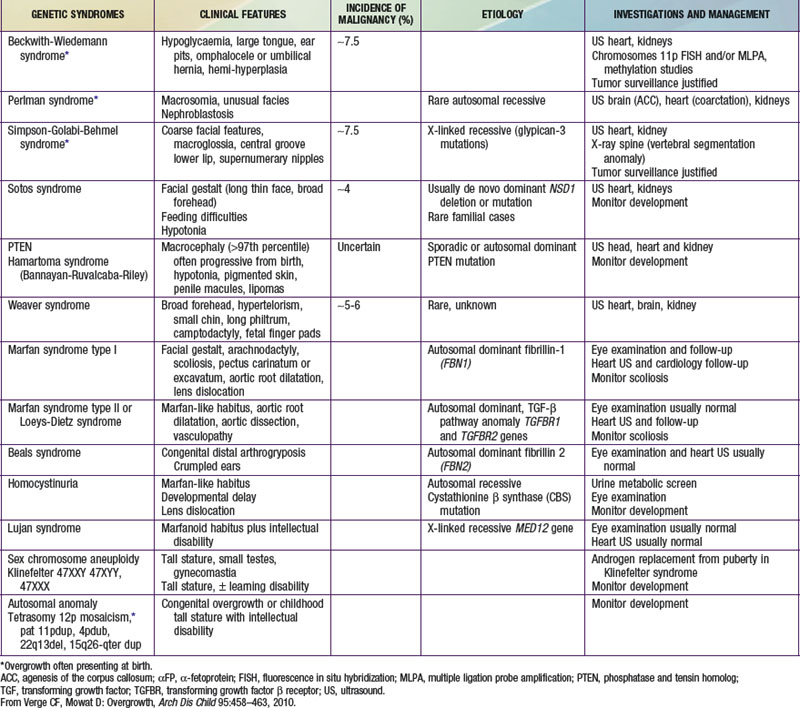
Table 554-1 lists the causes of tall stature in childhood and adolescence. Of those listed, the normal variant, familial or constitutional tall stature, is by far the most common cause. Almost invariably, a family history of tall stature can be obtained, and no organic pathology is present. The child is often taller than his or her peers throughout childhood and enjoys excellent health. The parent of the constitutionally tall adolescent might reflect unhappily upon his or her own adolescence as a tall teenager. There are no abnormalities in the physical examination, and the laboratory studies, if obtained, are negative. Additional features of overgrowth syndromes are noted in Table 554-2.
Klinefelter syndrome (XXY syndrome) is a relatively common (1 : 500-1000 live male births) abnormality associated with tall stature, learning disabilities (including requirement for speech therapy), gynecomastia, and decreased upper to lower body segment ratio. Affected boys can have hypotonia, clinodactyly, and hypertelorism. The testes are invariably small, although androgen production by Leydig cells is often in the low-normal range. Spermatogenesis and Sertoli cell function are defective, and infertility results. Other genital abnormalities including relatively small phallus, hypospadias, and cryptorchidism may be present.
XYY syndrome is associated with tall stature, problems in motor and language development, and possible antisocial behavior.
Marfan syndrome is an autosomal dominant connective tissue disorder consisting of tall stature, arachnodactyly, thin extremities, increased arm span, and decreased upper to lower body segment ratio (Chapter 693). Additional abnormalities include ocular abnormalities (e.g., lens subluxation), hypotonia, kyphoscoliosis, cardiac valvular deformities, and aortic root dilatation.
Homocystinuria is an autosomal recessive inborn error of amino acid metabolism, caused by a deficiency of the enzyme cystathionine synthetase. It is characterized by mental retardation when untreated, and many of its clinical features resemble Marfan syndrome, particularly ocular manifestations (Chapter 79). Hyperthyroidism in adolescents is associated with rapid growth but normal adult height. It is almost always caused by Graves disease and is much more common in girls (Chapter 562).
Exogenous obesity is a common condition in adolescence and may be associated with rapid linear growth and early onset of puberty. Adult height is typically normal.
The purpose of the diagnostic evaluation of tall stature is to distinguish the commonly occurring normal variant constitutional variety from the rare pathologic conditions. Often, when the history suggests familial tall stature and the physical examination is entirely normal, no laboratory tests are indicated. It is valuable to obtain a bone age radiograph to be able to predict adult height, which serves as a basis for discussions with the family and for management decisions. If the history suggests any of the aforementioned disorders or the physical examination reveals abnormalities, additional laboratory tests should be obtained. Insulin-like growth factor-1 (IGF-1) and IGF binding protein-3 (IGFBP-3) are excellent screening tests for GH excess and can be verified with a glucose suppression test. Laboratory evidence of GH excess mandates MRI evaluation of the pituitary. Chromosome analysis is useful in boys, especially when the ratio of upper to lower body segment is decreased or when developmental disability is present, to rule out Klinefelter syndrome. If Marfan syndrome or homocystinuria is suspected from the physical examination, referral to a cardiologist and an ophthalmologist should be made. Thyroid function tests are useful to diagnose or rule out hyperthyroidism when this disorder is suspected.
Precocious puberty, whether mediated centrally (increased gonadotropin secretion) or peripherally (increased secretion of androgens, estrogens, or both), results in accelerated linear growth during childhood, mimicking the pubertal growth spurt. Because skeletal maturation is also advanced, adult height is often compromised. The diagnostic evaluation and management of precocious puberty are discussed in Chapter 556.
Although delayed puberty may be associated with short stature in childhood, as with constitutional delay, failure to eventually enter puberty and complete sexual maturation can result in sustained growth during adult life, with ultimate tall stature. The report of tall stature with open epiphyses resulting from a mutation of the estrogen receptor in a man with normal male sexual maturation underscores the fundamental role of estrogen in promoting epiphyseal fusion and termination of normal skeletal growth. Aromatase deficiency leads to tall stature through similar pathways. Furthermore, androgen insensitivity is associated with tall stature in girls, demonstrating a role for androgens in this process.