Fig. 6.1
(a) Pilocytic astrocytoma showing biphasic architecture, with both a loose, hypocellular pattern (right half) and a compact cellular pattern (left half). H&E, original magnification ×100. (b) Rosenthal fibers (arrows) scattered within the compact piloid component of a pilocytic astrocytoma. H&E, original magnification ×200. (c) Eosinophilic granular body (arrow) within a loose area in a pilocytic astrocytoma. H&E, original magnification ×200
The presence of microscopic foci of brain or subarachnoid space infiltration, occasional mitotic figures, isolated foci of necrosis, or microvascular proliferation does not necessarily indicate malignancy or poor survival [5]. Innocent degenerative changes such as nuclear atypia and pleomorphism associated with smudgy chromatin and nuclear pseudo-inclusions can occur and may lead to misdiagnosis of a malignant glioma. Other degenerative changes that can be seen include the presence of multinucleated giant cells (often referred to as “pennies on a plate”). Malignant transformation is rare and typically results following radiation therapy of this low-grade lesion [5].
Immunohistochemically, pilocytic astrocytomas have a low Ki67/MIB-1 proliferative index of 1–4 % (but sometimes higher) and stain for GFAP. When the location of these tumors limits adequate biopsies, it is often difficult histologically to differentiate them from other low-grade lesions such as diffuse astrocytomas (WHO grade II). Molecular testing in these cases may provide some aid, as the majority (70 %) of cerebellar pilocytic astrocytomas exhibit activation of the RAS/BRAF/MAPK pathway commonly due to BRAF duplication/fusion with the KIAA1549 gene, something less commonly seen in diffuse astrocytomas [6].
A potential diagnostic mimic of pilocytic astrocytoma is nonneoplastic reactive gliosis which can resemble a pilocytic astrocytoma due to increased numbers of astrocytes and the presence of Rosenthal fibers. Such so-called piloid gliosis classically occurs at the rim of a craniopharyngioma or a hemangioma where the surrounding brain tissue creates a dense gliotic background filled with Rosenthal fibers, but can occur around any long-standing or slowly expanding lesion. Biopsies from these areas may be misinterpreted as findings consistent with a pilocytic astrocytoma, underscoring the need for adequate surgical sampling.
Pilomyxoid Astrocytoma
This pediatric tumor entity shares many of the gross and microscopic features of pilocytic astrocytomas. Like pilocytic astrocytomas, these tumors tend to occur in midline structures, with a predominance for the suprasellar/hypothalamic region [7]. Their increased tendency to recur and less favorable prognosis have resulted in designating pilomyxoid astrocytoma as a distinct WHO grade II entity rather than simply as a rare morphologic variant of pilocytic astrocytoma [7].
Grossly, pilomyxoid astrocytomas are well-circumscribed, solid, gelatinous, or cystic lesions that are difficult to distinguish macroscopically from pilocytic astrocytoma. On microscopic examination however, unlike its close relative, pilomyxoid astrocytoma is a monophasic tumor in a predominantly myxoid background matrix (Fig. 6.2a). Embedded within the myxoid matrix are bland bipolar cells that have a tendency to form an angiocentric pattern (Fig. 6.2b). Other histological findings that may help in distinguishing this entity from pilocytic astrocytoma are its lack of protoplasmic cells, Rosenthal fibers or calcifications, and rare eosinophilic granular bodies. Neither immunohistochemistry nor molecular testing has been helpful thus far in distinguishing pilomyxoid from pilocytic astrocytoma.
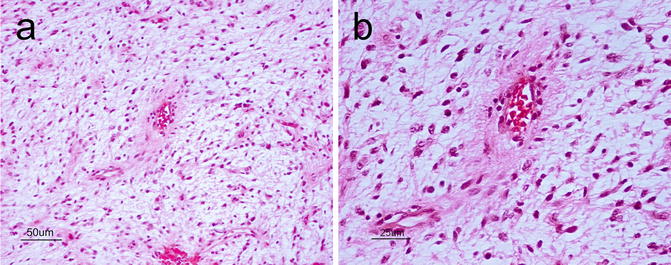
Fig. 6.2
(a) Pilomyxoid astrocytoma showing a monophasic architecture in a myxoid background. There is an impression of the angiocentric pattern at this low power that is better seen at higher power. H&E, original magnification ×200. (b), Angiocentric pattern of pilomyxoid astrocytoma. H&E, original magnification ×400
Angiocentric Glioma
This entity was added to the WHO 2007 classification as a WHO grade I lesion and carries a good prognosis [2, 8]. They are cortically based, typically occurring in the frontoparietal and temporal lobes (including the hippocampal region), and present clinically with seizures. Microscopically, angiocentric gliomas are variably infiltrative and characterized by monomorphous bipolar cells with a prominent angiocentric growth pattern (Fig. 6.3a). They also frequently display subpial palisading. As for most astrocytic neoplasms, GFAP is diffusely positive. There is also an intracytoplasmic dot-like pattern of staining with epithelial membrane antigen (EMA) (Fig. 6.3b).
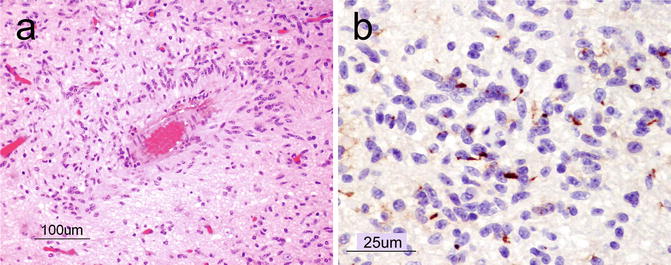
Fig. 6.3
(a) Angiocentric glioma showing elongated astrocytic tumor cells organizing around a blood vessel (angiocentric pattern). H&E, original magnification ×100. (b), A dot-like staining pattern with epithelial membrane antigen (EMA) immunohistochemistry is a characteristic feature of angiocentric glioma. Original magnification ×400
Diffuse Astrocytoma
Diffuse astrocytomas (WHO grade II) are infiltrating glial tumors that occur throughout the pediatric CNS. Unlike in adults, diffuse astrocytomas in children rarely progress to higher grade lesions. Grossly, this tumor forms an ill-defined and occasionally cystic mass that causes enlargement and distortion of the involved anatomical structures.
Microscopically, diffuse astrocytomas are composed of hypercellular sheets of cells with oval to elongate nuclei within a fibrillar background (Fig. 6.4a). The entrapment of nonneoplastic neurons (Fig. 6.4b) and features such as perineuronal, perivascular, subpial, and subependymal aggregates of neoplastic cells (the so-called secondary structures) are evidence of their infiltrative growth. The presence of atypical nuclei that are angular, hyperchromatic, and mildly pleomorphic can help differentiate these neoplasms from reactive astrocytes. Mitoses, however, are rare or absent. GFAP is the main immunohistochemical stain that highlights the neoplastic astrocytic cells. Neurofilament is helpful for highlighting the infiltrative nature of the tumor cells (Fig. 6.4c).
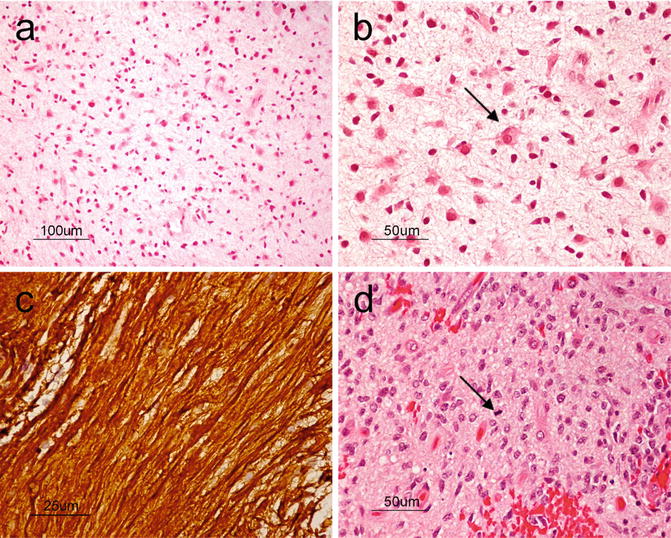
Fig. 6.4
(a) At low power, diffuse astrocytoma can be recognized as areas of hypercellularity within the brain. H&E, original magnification ×100. (b), On higher power, entrapped neurons (arrows) can be seen demonstrating the infiltrative nature of the tumor. H&E, original magnification ×200. (c), Neurofilament immunostain highlighting the infiltrative nature of the tumor. H&E, original magnification ×400. (d), Anaplastic astrocytoma showing markedly increased cellularity, nuclear pleomorphism, and mitosis (arrow). H&E, original magnification ×200
Morphological variants of diffuse astrocytomas include protoplasmic and gemistocytic variants. The former is a rare hypocellular, mucoid, and microcystic lesion that is predominantly composed of cells with small nuclei, low content of glial filaments, and scant GFAP expression. The gemistocytic variant shows a predominant population of cells with abundant eosinophilic cytoplasm, nuclei that are displaced to the periphery and strong, consistent GFAP expression.
Recently, a study applying whole-genome sequencing to low-grade gliomas found a high rate (53 %) of either intragenic duplication of the tyrosine kinase domain of the FGFR1 or rearrangements of MYB in WHO grade II diffuse gliomas [9]. In a similar analysis of grade II gliomas, 28 % were shown to have partial duplication of the transcription factor MYBL1 [10]. Given the availability of specific therapeutic inhibitors of such pathways, histopathological diagnosis with supplemental molecular analysis of tumors may one day offer more patient-specific treatments and prognostication for these tumors.
Anaplastic Astrocytoma
Anaplastic astrocytomas (WHO grade III) show increased cellularity, nuclear pleomorphism, and hyperchromasia when compared to WHO grade II diffuse astrocytomas. Grossly, it is difficult to differentiate between diffuse astrocytoma WHO grade II and anaplastic astrocytoma.
The presence of mitoses is a key distinguishing feature that favors a diagnosis of anaplastic astrocytoma over the lower grade astrocytic neoplasms already discussed (Fig. 6.4d). Though the finding of pyknotic nuclei is still consistent with this category, geographic areas of necrosis or microvascular proliferation are usually indicative of glioblastoma.
Glioblastoma
Glioblastoma (GBM) is a malignant astrocytic tumor (WHO grade IV) that can occur throughout the CNS. It is less common in the pediatric population than in adults. As its former name “glioblastoma multiforme” suggests, the tumor exhibits tremendous phenotypic heterogeneity from one region of the tumor to another. Grossly, it usually shows areas of necrosis, hemorrhage, peritumoral edema as well as occasional pseudocapsule formation, in addition to a yellowish discoloration from myelin breakdown. GBMs also exhibit variable architectural patterns and cellular morphologies at the microscopic level that include multinucleated giant cells, small cells, granular cells, and lipidized cells. In addition to sharing features such as hypercellularity, diffuse infiltration, pleomorphism, and mitoses with anaplastic astrocytoma, GBMs also show either necrosis, typically with pseudopalisading of tumor cells (Fig. 6.5a) or microvascular proliferation (Fig. 6.5b).
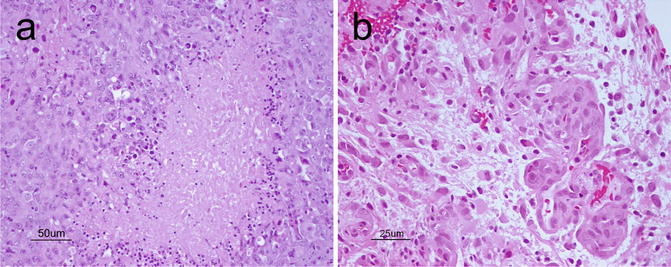
Fig. 6.5
(a), Classical palisading of the neoplastic cells around areas of necrosis. H&E, original magnification ×200. (b) vascular-endothelial proliferation. H&E, original magnification ×400
Exome sequencing of pediatric supratentorial/hemispheric GBMs has revealed differences in the biology that governs tumorigenesis between pediatric/young adult GBMs and those seen in adults. In the young age group somatic mutations in the H3.3-ATRX-DAXX chromatin remodeling pathway can be found. Mutations in the tumor suppressor TP53 are found in approximately half of GBMs and 86 % of GBMs with H3.3 or ATRX mutations. Mutations in H3.3 were also highly specific for GBMs when compared to low-grade lesions and thus provide additional tools to help differentiate between the different tumor grades in small biopsies [11]. Although extremely rare in the pediatric population, IDH1 mutations may be encountered in the adolescent age group [12].
Pleomorphic Xanthoastrocytoma
Pleomorphic Xanthoastrocytoma (PXA) is a relatively rare astrocytic, low-grade tumor (WHO grade II) with distinctive morphologic features. It is a supratentorial lesion that classically involves the superficial cortex and leptomeninges of the temporal lobe of children and young adults with medically refractory epilepsy. Grossly, PXAs are discrete lesions with leptomeningeal involvement and may be cystic lesions. Microscopically, as its name suggests, PXAs are composed of highly pleomorphic, fibrillary astrocytes, some of which may show lipidization (Fig. 6.6a). Large, bizarre, multinucleated cells are an additional feature. Reticulin positivity is a characteristic feature (Fig. 6.6b). Eosinophilic granular bodies, perivascular infiltration by lymphocytes, and a solid component within the subarachnoid space are key features that can help with diagnosis. Due to its significant pleomorphism, differentiation of PXA from more malignant astrocytic tumors such as anaplastic astrocytoma or GBM is critical given the difference in prognosis. In fact, even when PXAs show “anaplastic features” (5 or more mitoses per 10 high power fields) or necrosis, the prognosis still remains more favorable than that of high-grade gliomas [13]. The neoplastic cells in PXA are positive for S100 and variably positive for GFAP and CD34 [14]. There may also be focal areas with a neuronal phenotype suggesting a multipotential precursor cell as its cell of origin [15]. Approximately two-thirds of PXAs harbor the V600E BRAF mutation [16].
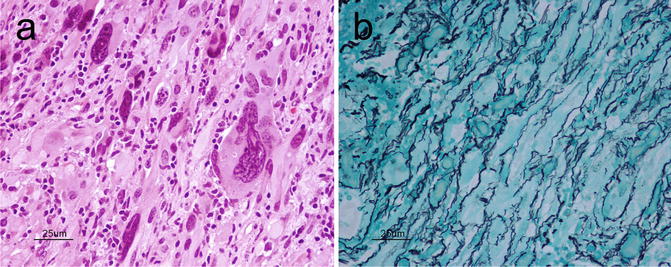
Fig. 6.6
(a) PXA showing pleomorphic fibrillary astrocytes, including large bizarre cells. H&E, original magnification ×400. (b), PXA typically exhibits an extensive reticulin network. Reticulin, original magnification ×400
Embryonal Tumors
Medulloblastoma
Medulloblastoma (WHO grade IV) is the most common malignant pediatric brain tumor. It characteristically occurs in the cerebellum and in the roof of the 4th ventricle, where it forms a well-circumscribed, soft, gray mass presenting as cerebellar dysfunction and obstructive hydrocephalus with cranial nerve findings. It has a tendency to infiltrate the leptomeninges and form spinal “drop metastases” via the CSF.
Histologically, it is divided into classic medulloblastoma (majority of cases) and less common variants that include desmoplastic/nodular medulloblastoma, medulloblastoma with extensive nodularity, anaplastic medulloblastoma, and large cell medulloblastoma.
As discussed in Chap. 12, medulloblastomas can also be divided into four distinct molecular subgroups with some enrichment for the histological types. Here we will discuss the histologic variants.
The classic medulloblastoma subtype makes up approximately 70 % of all medulloblastomas and forms sheets of small round blue cells (high nucleus-to-cytoplasmic ratio) containing variable numbers of Homer-Wright (neuroblastic) rosettes characterized by a fibrillary core (Fig. 6.7a,b) and rarely differentiating ganglioneuronal elements. Medulloblastomas usually exhibit a high mitotic rate, numerous apoptotic bodies, and small areas of necrosis. The tumor cells show positivity for neuronal immunohistochemical markers such as synaptophysin, NeuN, anti-Hu, and neuron-specific enolase in addition to focal positivity with glial markers like GFAP [17].
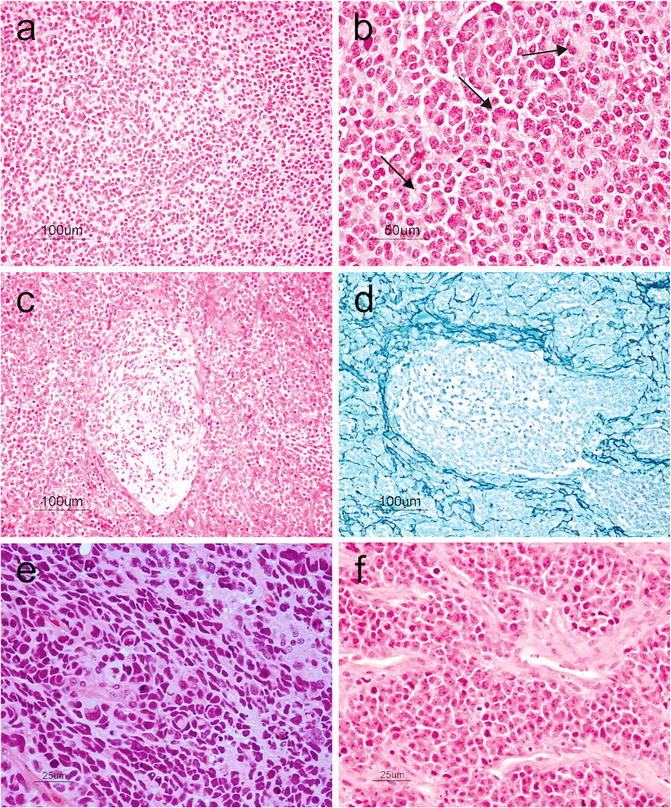
Fig. 6.7
(a) Small round blue cells characteristic of medulloblastoma. H&E, original magnification ×100. (b) Tumor cells arranged in Homer-Wright rosettes (arrows). H&E, original magnification ×200. (c), Desmoplastic medulloblastoma showing pale nodular areas in between more cellular internodular regions. H&E, original magnification ×100. (d) Reticulin-rich internodular regions in desmoplastic medulloblastoma. Reticulin, original magnification ×100. (e) Anaplastic medulloblastoma showing markedly pleomorphic cells, cell molding, and prominent apoptosis. H&E, original magnification ×400. (f) Large cell medulloblastoma showing large anaplastic cells with prominent nucleoli and vesicular chromatin. Note the prominent apoptosis. H&E, original magnification ×400
Nodular/desmoplastic medulloblastoma is the second most common histopathological variant of medulloblastomas. Grossly, it has a firm consistency and unlike the classic form that tends to occur in the vermis, it arises in the cerebellar hemispheres. Microscopically, pale zones showing neurocytic differentiation and apoptosis are surrounded by more cellular zones that are reticulin-rich and highly proliferative (Fig. 6.7c,d). The differentiated nodules may be highlighted using neuronal markers such as synaptophysin. Nodular/desmoplastic medulloblastomas are associated with Gorlin syndrome and activation of the sonic hedgehog pathway [17].
Medulloblastoma with extensive nodularity is a rare variant, usually seen in infants, with a favorable prognosis. It is qualitatively differentiated from the nodular/desmoplastic type variant by having a predominant lobular architecture with large elongated reticulin-free zones that contain small round neurocytic cells in a fibrillary background. The internodular component of reticulin-rich areas that is commonly seen in desmoplastic medulloblastoma is largely reduced [18].
Anaplastic/large cell medulloblastomas are thought to be more aggressive variants. Anaplastic medulloblastomas show marked nuclear enlargement, nuclear pleomorphism, nuclear molding, high mitotic count, prominent necrosis, and apoptosis (Fig. 6.7e). Large cell medulloblastoma has discohesive, monomorphic large cells with round nuclei, open chromatin, and prominent nucleoli (Fig. 6.7f). The features of “anaplastic” and “large cell” may coexist in the same tumor [2].
Other variants include medulloblastomas with melanotic or myogenic differentiation that can be highlighted by melanocytic and myogenic markers, respectively. Although histologically distinctive, these variants do not have any known prognostic significance at this time [2].
Even among histologically similar medulloblastomas, genomic approaches have helped further subclassify these tumors into clinical and biologically distinct groups. Four subgroups are currently recognized: WNT, SHH, Group 3, and Group 4. Not only will these groups provide better prognostic information for patients, but these also help stratify tumors into classes driven by distinct molecular pathways, which have the potential to be translated clinically into more patient-specific therapies [19]. These classes are discussed in more detail in Chap. 12.
Supratentorial Primitive Neuroectodermal Tumor
Supratentorial Primitive Neuroectodermal Tumor (sPNETs) (WHO grade IV) are tumors morphologically similar to medulloblastomas but that reside in the cerebral hemispheres rather than the cerebellum of children and adolescents. Compared to medulloblastomas, sPNETs are less frequently encountered and carry a worse prognosis. This tumor is mostly situated deep within the hemisphere. Usually, they are soft and pink-red to purple in color with or without cystic changes or hemorrhages. Microscopically, they resemble medulloblastomas exhibiting sheets of poorly differentiated small round blue cells with hyperchromatic nuclei and a high nucleus-to-cytoplasmic ratio (Fig. 6.8a). Homer-Wright rosettes are found with variable frequency. Areas of necrosis, abundant mitoses, and apoptosis can be seen [2]. Immunohistochemistry may demonstrate areas of divergent differentiation including neuronal, glial or, more rarely, mesenchymal. When large ganglion cells are seen disbursed throughout neuroblastic differentiation, the term ganglioneuroblastoma may be used [2].
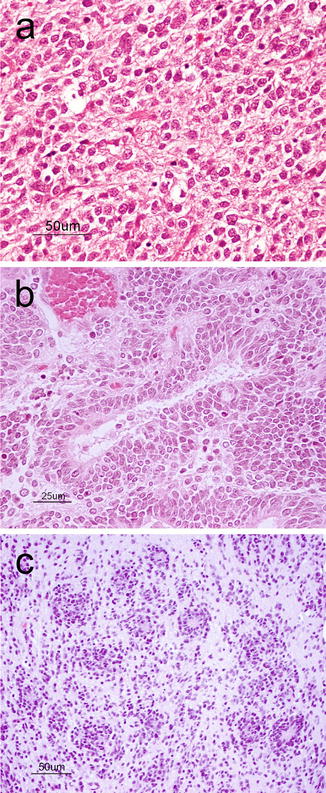
Fig. 6.8
(a) sPNET showing a cellular neoplasm with hyperchromatic, anaplastic nuclei, and numerous apoptotic bodies. H&E, original magnification ×200. (b) Medulloepithelioma showing the characteristic tubular structures mimicking the primitive neural tube. H&E, original magnification ×400. (c) Embryonal tumor with abundant neuropil and true rosettes demonstrating the biphasic architecture with hypercellular regions abutting hypocellular fibrillary regions. H&E, original magnification ×200
Similar and very rare primitive embryonal tumors include those with predominant neural tube formation termed medulloepithelioma (Fig. 6.8b). A newly described variant is the so-called “embryonal tumor with abundant neuropil and true rosettes” (Fig. 6.8c) that shows focal areas of hypercellularity, broad bands of neuropil and rosettes with slit-like lumens and has an extremely poor outcome [20]. This tumor is characterized by frequent amplifications at chromosome 19q13 [21].
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


