Hirschsprung Disease
Hirschsprung disease (HD), also known as ‘congenital megacolon’ is characterized by the absence of ganglion cells in the myenteric and submucosal plexuses of the intestine. The first known description of this condition was by ancient Hindu surgeons in the Shushruta Samheta,1 and the first descriptions in the modern medical literature were from the 17th century.2 In 1887, Harald Hirschsprung, a pediatrician from Copenhagen, described two cases of the condition that ultimately bore his name.3 At that time most children with congenital megacolon died from malnutrition and enterocolitis. As the underlying pathological basis of the disease was unknown, surgeons removed the massively dilated proximal bowel and created a colostomy. Attempts at re-anastomosis were uniformly unsuccessful.4
Although the absence of ganglion cells in the distal colon of a child with HD was first noted by Tittel in 19015 and subsequent publications repeated this observation, it took many decades for clinicians caring for these children to become aware. The first recognition of aganglionosis by a surgeon as the cause of congenital megacolon was by Ehrenpreis in 1946.6 This was followed in 1949 by Swenson’s first description of a reconstructive operation for HD.7 Although Swenson’s operation was originally performed without a colostomy, technical difficulties in small infants and the debilitated and malnourished state in which many children presented were reasons most surgeons adopted a multi-staged approach with colostomy as the initial step.8 In recent years, improvements in operative technique and earlier diagnosis have resulted in an evolution toward one-stage and minimal access procedures. These advances have resulted in significantly improved morbidity and mortality in infants with HD.
Incidence and Spectrum of Disease
A number of syndromes are associated with HD including trisomy 21, congenital central hypoventilation syndrome, Goldberg–Shprintzen syndrome, Smith–Lemli–Opitz syndrome, neurofibromatosis, and neuroblastoma (Box 34-1).
Etiology and Genetic Basis of Disease
Ganglion cells are derived from the neural crest. By 13 weeks post-conception, the neural crest cells have migrated from proximal to distal through the gastrointestinal tract, after which they differentiate into mature ganglion cells.9 There are two main theories why this process is disturbed in children with HD. The first possibility is that the neural crest cells never reach the distal intestine due to early maturation or differentiation into ganglion cells. Data supporting this theory come from animal models showing spontaneous aganglionosis10,11 and from studies of normal neural crest cell migration performed in chick embryos and human fetuses.12,13 The second possibility is that the neural crest cells reach their destination, but fail to survive or differentiate into ganglion cells due to an inhospitable microenvironment.14,15 It is likely that HD is actually a heterogeneous group of diseases with multiple genetic causes and etiologies.
A genetic basis for HD has long been suspected because of the presence of a family history in many cases and the known association with trisomy 21 and other genetically based conditions. Over the past two decades, an increasing number of researchers have made significant progress in identifying and elucidating the complex array of genetic mutations and mechanisms responsible for this disease.16–18 The first and most common gene to be identified is the RET proto-oncogene, which encodes a tyrosine kinase receptor. Many mutations of this gene and related genes, such as neurturin and glial cell line-derived neurotrophic factor (GDNF), have been described. It remains unclear how these mutations result in aganglionosis, but there is evidence that early neuronal cell death may be a prominent mechanism.19,20 RET abnormalities are most commonly found in patients with familial and long-segment involvement. Mutations in the endothelin family of genes, particularly endothelin-3 and the endothelin-B receptor, are also commonly associated with HD. Many of these children have other neurocristopathies such as dysfunction of melanocytes, congenital deafness, central hypoventilation, and neuroblastoma. From animal models, there is evidence that mutations in the endothelin and SOX-10 genes may produce early maturation or differentiation of neural crest cells, which decreases the number of available progenitor cells and prevents the neural crest cells from migrating any further.21,22 Other genes associated with HD include S1P1 (now known as ZFHX1B), Phox2B, and the Hedgehog/Notch complex.23
Clinical Presentation and Diagnosis
Prenatal diagnosis of HD is rare, and is usually due to total colonic disease resulting in ultrasound (US) findings of fetal intestinal obstruction.24 Most affected patients present during the neonatal period with abdominal distension, bilious vomiting, and feeding intolerance. Delayed passage of meconium beyond the first 24 hours is present in approximately 90%. Occasionally, cecal or appendiceal perforation may be the initial event.25 Plain radiographs characteristically show dilated bowel loops throughout the abdomen. The next step is a water-soluble contrast enema. The pathognomonic finding of HD is a transition zone between the normal and aganglionic bowel (Fig. 34-1), although approximately 10% of neonates with HD may not have a demonstrable radiological transition zone.26 Occasionally, false positive studies occur.27 It is also important to obtain a plain radiograph 24 hours later. Retention of the contrast is very suggestive for HD (Fig. 34-2). It is also important to use a water-soluble material as the enema potentially may be a definitive treatment for other conditions in the differential diagnosis, such as meconium ileus and meconium plug syndrome. Once the diagnosis of HD is suspected, the diagnosis must be confirmed by rectal biopsy, which in the neonate can be done at the bedside without sedation and using a suction technique.
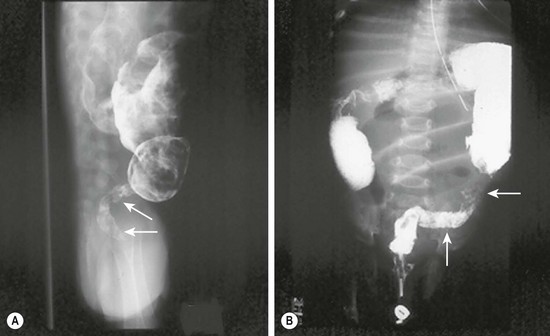
FIGURE 34-1 These two barium enema examinations in different infants demonstrate Hirschsprung disease. The aganglionic rectum (arrows) in both studies is small and contracted. The proximal ganglionic colon is dilated. A transition zone between the aganglionic and ganglionic colon is nicely seen in both studies.
FIGURE 34-2 Retention of contrast is seen on this post-evacuation film which was obtained 24 hours after the contrast enema.
Patients presenting later in childhood have chronic severe constipation. As constipation is common in children, it can be difficult to differentiate HD from more common causes. Clinical features pointing to the diagnosis include delayed passage of meconium at birth, failure to thrive, abdominal distention, and dependence on enemas without significant encopresis.28 Although a contrast enema usually demonstrates a transition zone in older children, a false negative study may be due to massive rectal distension in combination with a very short aganglionic segment. Reversal of the usual rectosigmoid ratio and retention of contrast on a 24-hour postevacuation film also support the diagnosis. Anorectal manometry is another useful screening technique, in which the presence of a recto-anal inhibitory reflex (reflex relaxation of the internal anal sphincter in response to balloon distension of the rectum) essentially rules out HD (Fig. 34-3). In older children, the suction rectal biopsy may be less reliable because of a higher risk of sampling error. Full-thickness biopsies, usually under general anesthesia, may be necessary in these patients.
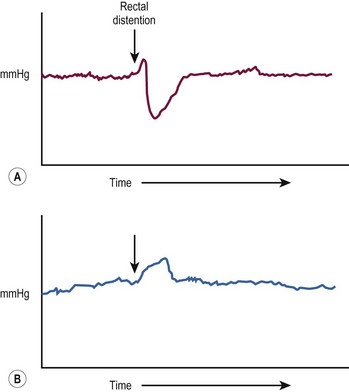
FIGURE 34-3 (A) In the child without Hirschsprung disease undergoing anorectal manometry, the recto-anal inhibitory reflex is normal. Note the drop in the internal sphincter pressure with rectal distention. (B) A child with Hirschsprung disease is seen to have abnormally increased contraction of the anal canal and no relaxation of the internal sphincter with rectal distention. (The arrow points to the initiation of rectal distention in both A and B.)
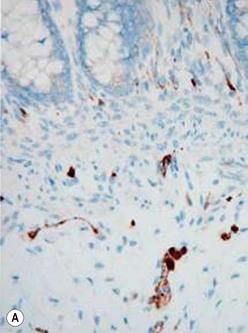
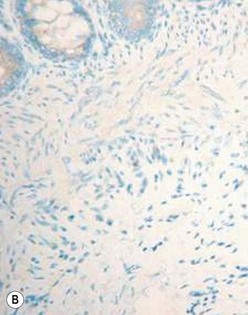
The gold standard for the diagnosis is the absence of ganglion cells in the submucosal and myenteric plexuses on histological examination (Fig. 34-4A). Most patients will also have evidence of hypertrophied nerve trunks (Fig. 34-4B), although this finding is not always present, particularly in children with total colonic disease or a very short aganglionic segment. As there is normally a paucity of ganglion cells in the area 0.5–1.0 cm above the dentate line, the rectal biopsy should be taken at least 1.0–1.5 cm above it. However, a biopsy too proximal may miss a short aganglionic segment. In addition to hematoxylin and eosin, many pathologists also stain for acetylcholinesterase, which has a characteristic pattern in the submucosa and mucosa in children with HD (Fig. 34-5). Recently, it has been shown that immunochemical staining for calretinin is almost always absent in patients with HD (Fig. 34-6).29
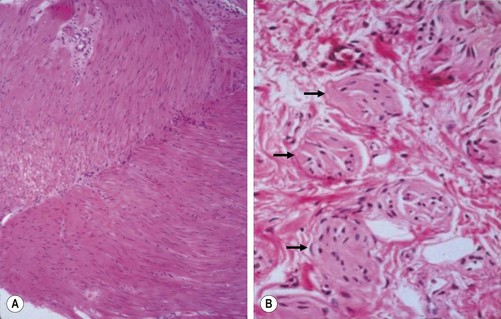
FIGURE 34-4 Histological findings in children with Hirschsprung disease. (A) Absence of ganglion cells in the myenteric plexus. (B) Hypertrophied nerve trunks are marked with arrows.
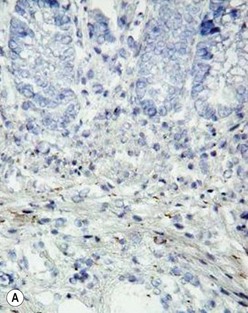
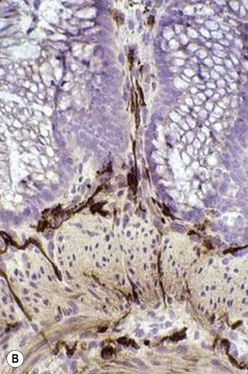
FIGURE 34-5 Cholinesterase staining in (A) normal colon and (B) colon affected by Hirschsprung disease.
Preoperative Preparation
Some physicians have advocated nonoperative long-term management of short segment HD using enemas and laxatives. Others have suggested that simple myectomy may be adequate.30 However, these approaches do not provide a good quality of life for most infants and children with HD, and most pediatric surgeons recommend a pull-through procedure even for short segment disease.
Surgical Management
The goals of surgical management for HD are to remove the aganglionic bowel and reconstruct the intestinal tract by bringing the normally innervated bowel down to the anus while preserving normal sphincter function. The most commonly performed operations are the Swenson, Duhamel and Soave procedures, although a number of other operations, such as the Rebhein and State procedures, have been described and are still performed in some centers.31 As there are very few prospective studies comparing operations, the best operation for an individual patient is the one that the surgeon has been trained to do and does frequently.
Although Swenson’s operation was initially developed as a one-stage procedure, the relatively high incidence of stricture, leak, and other adverse outcomes led to the adoption of a routine preliminary colostomy, with definitive pull-through performed three to 12 months later.32 In the 1980s, a number of surgeons reported series of single-stage pull-through operations even in small infants.33,34 Over the following ten to 15 years, many reports suggested that a one-stage approach was safe, avoided the morbidity of stomas in infants, and was more cost effective.35–37 However, a stoma may still be needed for infants and children with severe enterocolitis, perforation, malnutrition, or massively dilated proximal bowel, and in situations when it is not possible to reliably identify the transition zone on frozen section.
Swenson Procedure
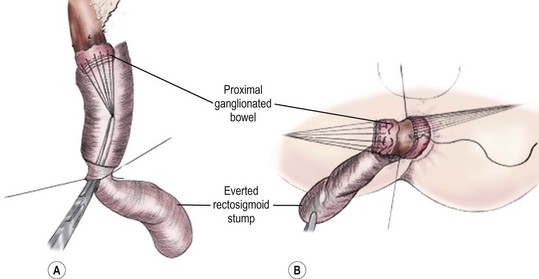
The goal of the Swenson pull-through is to remove the entire aganglionic colon, with an end-to-end anastomosis above the anal sphincter. The operation was originally performed via a laparotomy, with the anastomosis being performed from the perineum after everting the aganglionic rectum (Fig. 34-7). It is important to keep the dissection in the correct plane along the rectal wall to avoid injury to the deep pelvic nerves, vessels and other structures such as the vagina, prostate, vas deferens, and seminal vesicles. Despite the theoretical risks inherent in the deep pelvic dissection, long-term functional outcomes after the Swenson procedure are excellent.38
Soave Procedure
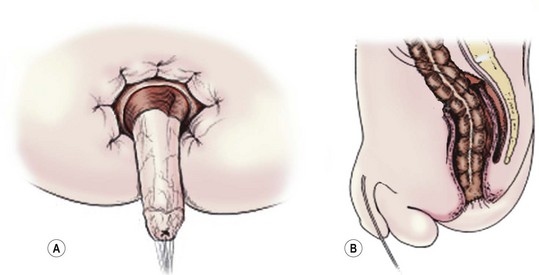
The Soave procedure, subsequently modified by Boley, was designed to avoid the risk of injury to important pelvic structures by performing a submucosal endorectal dissection and positioning the pull-through bowel within an aganglionic muscular ‘cuff’ (Fig. 34-8). Despite concerns by some that the Soave procedure may result in long-term constipation due to incomplete excision of the aganglionic rectum,39 most late follow-up studies have reported similar outcomes between the Soave and Swenson operation.40
Duhamel Procedure
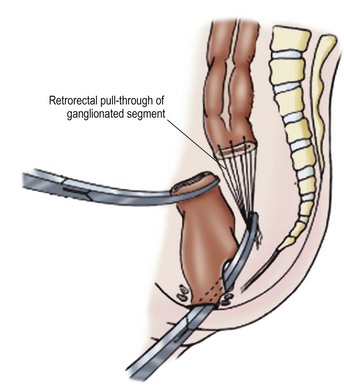
The Duhamel procedure involves bringing the normal colon down through the bloodless plane between the rectum and sacrum, and joining the two walls with a linear stapler to create a new lumen which is aganglionic anteriorly and normally innervated posteriorly (Fig. 34-9). The Duhamel operation is felt by many surgeons to be easier and safer than the Swenson or Soave procedures. Also, it results in a very large anastomosis, which reduces the risk of stricture. Reported longterm results of the Duhamel procedure have been similar to those with the other two operations, although recent studies suggest that outcomes from the Duhamel procedure are inferior to those of the transanal pull-through.41,42
Laparoscopic Pull-through
Georgeson first described the laparoscopic approach for HD in 1995.43 With this technique, a biopsy is initially performed to identify the transition zone, the rectum is mobilized below the peritoneal reflection, and a short mucosal dissection is performed through a perineal approach (Figs 34-10 to 34-14). The rectum is then prolapsed through the anus and the anastomosis performed from below. This procedure is associated with a shorter time in the hospital, and both early and mid-term results appear to be equivalent to those reported for the other procedures.44 Laparoscopic approaches have been also described for the Duhamel and Swenson operations, with excellent short term results.45,46
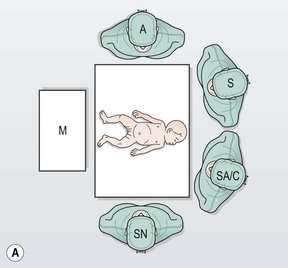
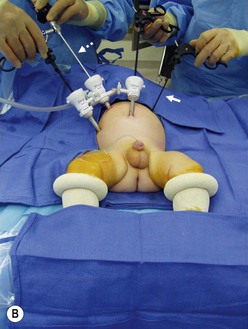
FIGURE 34-10 (A) The surgeon (S) and surgical assistant/camera holder (SA/C) stand above the patient’s head with the monitor (M) positioned beyond the infant’s feet. The scrub nurse (SN) can be positioned according to the surgeon’s preference, although being positioned at the foot of the operating table appears to be ideal. A, anesthesiologist. (B) The photograph shows port placement for this operation. Usually three or four ports are required. The umbilical port is inserted using an open technique, and the other ports are introduced under direct visualization. The telescope (dotted arrow) is placed through the 5 mm port in the right upper abdomen. The surgeon’s two primary working ports are the umbilical port for the left hand and the right lower abdominal port for the right hand. A retracting instrument (solid arrow) is often helpful and can be inserted through a stab incision in the infant’s left upper abdomen. A urinary catheter has been introduced to help decompress the bladder. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
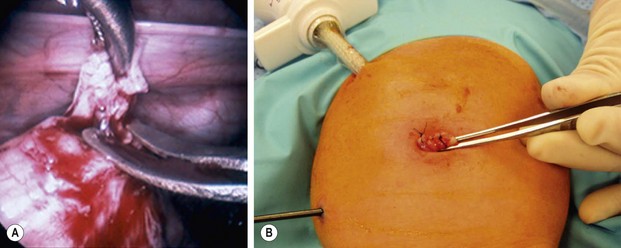
FIGURE 34-11 (A) An intracorporeal biopsy is being performed on the sigmoid colon. A fine-tipped grasping forceps has been used to grasp the biopsy site, and Metzenbaum scissors are used to obtain the biopsy specimen. (B) This biopsy was performed through the umbilical incision. One port and another instrument have been introduced through the infant’s abdominal wall. A site on the colon for the biopsy was visualized and delivered just under the umbilical cannula. The umbilical cannula was removed, and this portion of the colon was grasped and exteriorized. An extracorporeal biopsy was obtained and the biopsy site was closed. This is an alternative means for obtaining the biopsy. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
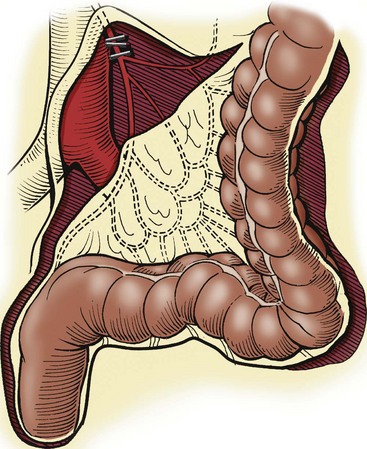
FIGURE 34-12 When the transition zone is proximal to the mid-sigmoid colon, a pedicled colon flap must be developed for the endorectal pull-through. In this situation, the pull-through colon will derive its vascular supply from the marginal artery. Therefore, to mobilize the descending colon and splenic flexure, it is necessary to ligate and divide either the inferior mesenteric artery just distal to its origin from the aorta (as seen in this drawing) or the left colic artery just after it arises from the inferior mesenteric artery. By ligating these vessels at these sites, the arterial supply through the marginal artery is not compromised. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
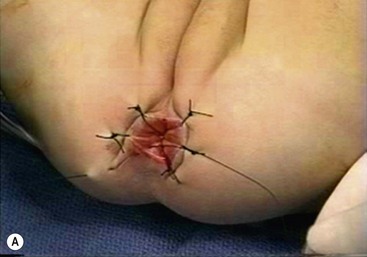
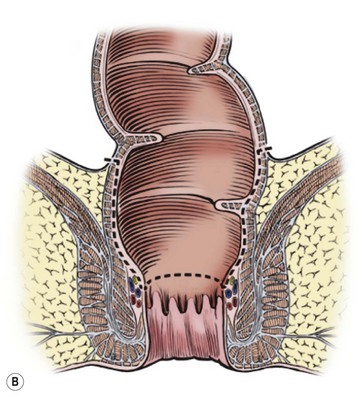
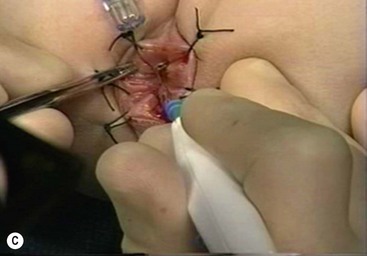
FIGURE 34-13 (A) The perineal dissection begins with the placement of circumferential 2-0 silk traction sutures from the dentate line to the perineum 2 to 3 cm outward from the anus. (B, C) A needle-tipped electrocautery is used to circumferentially incise the rectal mucosa approximately 5 mm proximal to the anal columns. Fine silk traction sutures are then placed in the rectal mucosa to help retract the mucosa during circumferential dissection. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
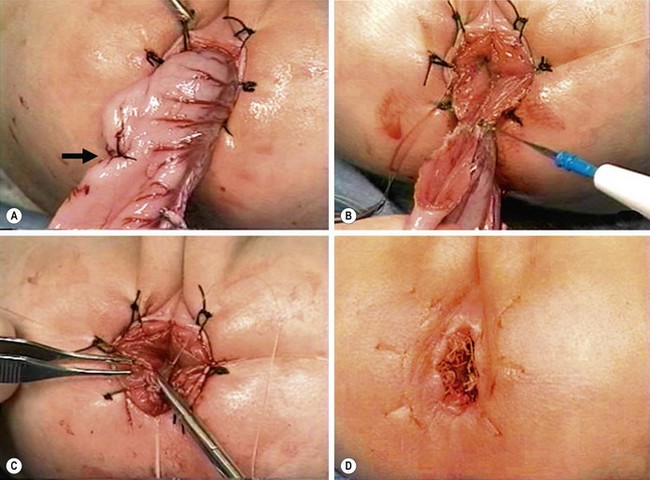
FIGURE 34-14 (A) The muscular cuff of the rectum has been divided and the ganglionic colon has been exteriorized through the anal canal. Note that the anastomosis will be performed proximal to the biopsy site (arrow). (B) The pull-through colon is being completely transected above the biopsy site and made ready for the coloanal anastomosis. (C) The anastomosis is being performed with interrupted 4-0 absorbable sutures. (D) The everting stay sutures have been cut, allowing the anastomosis to retract cephalad. (From Morowitz MJ, Georgeson KE. Laparoscopic assisted pull-through for Hirschsprung’s disease. In: Holcomb GW, Georgeson KE, Rothenberg SS, editors. Atlas of Pediatric Laparoscopy and Thoracoscopy. Philadelphia: Elsevier; 2008. p. 101–108. Reprinted with permission.)
Transanal (Perineal) Pull-through
The transanal approach was first described by de la Torre in 199847 and by Langer in 1999,48 and has been adopted and reported by an increasing number of surgeons. The operation can be performed in the prone or lithotomy positions.49 A mucosal incision is made 0.5–1.0 cm above the dentate line, depending on the size of the child, and the mucosa is stripped from the underlying muscle as in the Soave operation. The length of the submucosal dissection varies according to surgeon, although a shorter rectal cuff may be associated with a lower incidence of enterocolitis and the need for dilatation.50 Some surgeons do not perform any submucosal dissection, and effectively perform a transanal Swenson procedure.51
The rectal muscle is incised circumferentially, and the dissection is continued on the rectal wall, dividing the vessels as they enter the rectum. The entire rectum and part of the sigmoid colon can be delivered through the anus. When the transition zone is reached, the anastomosis is performed from below (Fig. 34-15). In patients with a more proximal transition zone (usually above the proximal sigmoid colon), laparoscopy or a small umbilical incision is needed to mobilize the left colon and/or splenic flexure to achieve an adequate length of ganglionated colon for pull-through.52 A transanal approach can also be used if the patient has already had a colostomy by using the stoma as the end of the pull-through bowel and performing the rectal excision using the transanal approach.