Chapter 157 Hereditary Periodic Fever Syndromes
The most common hereditary periodic fever disorders are familial Mediterranean fever (FMF), tumor necrosis factor (TNF) receptor–associated periodic syndrome (TRAPS), and hyperimmunoglobulinemia D syndrome (HIDS) (Table 157-1). The cryopyrin-associated periodic syndromes (CAPS) include Muckle-Wells syndrome (MWS), familial cold autoinflammatory syndrome (FCAS) (also known as familial cold urticaria [FCU]), and chronic infantile neurologic cutaneous and articular (CINCA) disease (also known as neonatal-onset multisystem inflammatory disease [NOMID]). A syndrome called pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA) and Blau syndrome (known also as familial juvenile systemic granulomatosis) have now been added to this group. Secondary amyloidosis (AA amyloidosis) is a complication in all of these periodic fever disorders, although it is less commonly reported with HIDS. FMF and HIDS are autosomal recessive diseases, whereas TRAPS, PAPA, and Blau syndrome are autosomal dominant conditions. The diagnosis of each of these entities depends on the clinical features and the genetic confirmation (see Table 157-1). Another periodic fever syndrome is periodic fever, aphthous stomatitis, pharyngitis, and cervical adenitis (PFAPA), but it is not clear yet whether PFAPA is an autoinflammatory syndrome (see Table 157-1). Among the conditions that are not in the category of periodic fever and that have been classified as autoinflammatory diseases are Crohn disease, Behçet diseases, early-onset childhood sarcoidosis, systemic juvenile idiopathic arthritis (JIA), and chronic recurrent multifocal osteomyelitis (known also as Majeed syndrome) (Table 157-2).
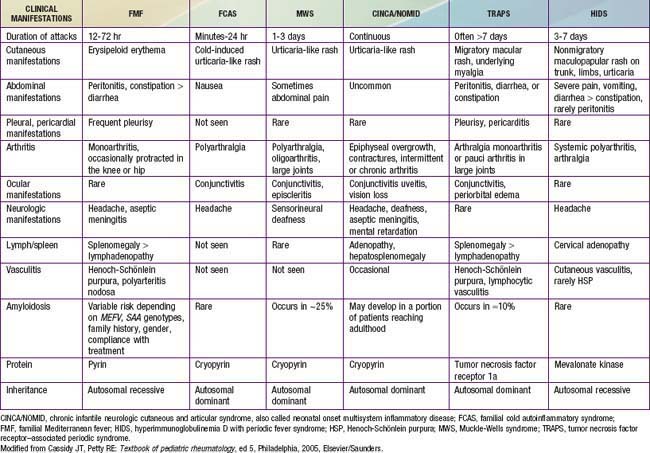
Table 157-2 RECURRENT OR PERIODIC FEVER SYNDROMES IN CHILDREN
INFECTIOUS DISEASES
RHEUMATIC DISEASES
HEREDITARY AUTOINFLAMMATORY SYNDROMES
CYCLIC HEMATOPOIESIS
IDIOPATHIC CONDITIONS
Periodic fever with aphthous stomatitis, pharyngitis, and adenitis (PFAPA)
From Cassidy JT, Petty RE: Textbook of pediatric rheumatology, ed 5, Philadelphia, 2005, Elsevier/Saunders.
Familial Mediterranean Fever
FMF is an autosomal recessive disorder characterized by brief, acute, self-limited episodes of fever and polyserositis that recur at irregular intervals and are associated with development of AA amyloidosis (Chapter 158).
Pathogenesis
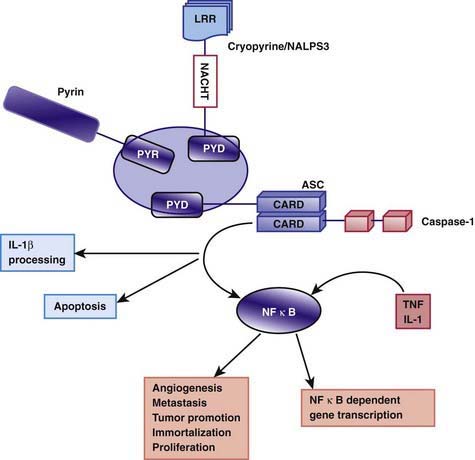
The exact pathogenesis of the acute episodes of FMF is unknown. Between episodes, patients with FMF have increased serum levels of interferon-γ and enhanced production of other proinflammatory cytokines, such as TNF-α, IL-1β, IL-6, and IL-8, in circulating leukocytes. Pyrin/marenostrin is a member of the death domain superfamily and consists of 4 different functional domains that interact with other proteins. Of particular interest is the domain known as the pyrin domain (PYD), a 92–amino acid N-terminal domain shared by several proteins that are involved in the regulation of the inflammatory response and apoptosis. Pyrin acts as an anti-inflammatory factor by inhibiting processing of pro–IL-1β cytokine to the active form. This inhibition normally takes place through interactions with a caspase recruitment domain (ASC) and NF-κB. It has been suggested that normally pyrin inhibits the binding of ASC to caspase-1 in a competitive manner. The C-terminal domain of the pyrin molecule interacts with caspase-1, leading to inhibition of IL-1β production. It is speculated that the defective (or mutated) pyrin found in patients with FMF is functionally inactive, allowing binding of ASC to caspase-1 to take place. As a consequence, stimulation of IL-1β processing and secretion occur, resulting in increased IL-1β levels that are responsible for the uncontrolled inflammation (Fig. 157-1). Another possibility that was previously more popular is based on the finding of C5a inhibitor (inactivating enzyme) deficiency in peritoneal and synovial fluids of patients with FMF. C5a is a fragment of complement, an anaphylatoxin, and a potent chemotactic agent (Chapter 127). Normally, C5a inhibitor neutralizes the small amounts of C5a released into serosal cavities before they precipitate overt inflammation. The hypothesis is that a deficiency of C5a inhibitor, which is a consequence of pyrin/marenostrin dysfunction in patients with FMF, allows further accumulation of C5a, leading to the acute attack. Further understanding of pyrin/marenostrin functions will shed light on aspects of FMF pathogenesis that are not yet fully understood.