Hereditary Gynecologic Cancers
KAREN H. LU  ANDREW BERCHUCK
ANDREW BERCHUCK  NOAH D. KAUFF
NOAH D. KAUFF
INTRODUCTION
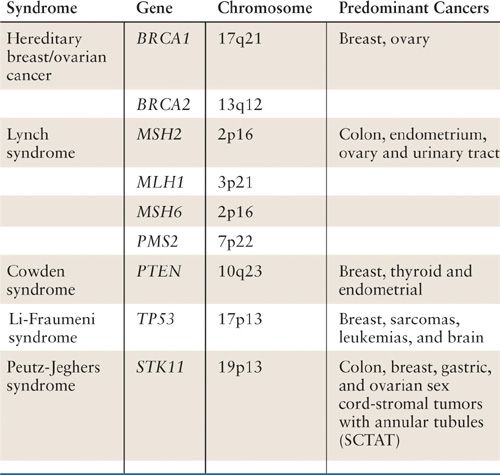
Identifying a woman with gynecologic cancer as having a hereditary cancer syndrome has tremendous implications for both the patient herself and for her family members. Hereditary Breast and Ovarian Cancer syndrome, caused by germline mutations in BRCA1 or BRCA2, as well as Lynch syndrome, caused by germ-line mutations in MLH1, MSH2, MSH6, or PMS2, are the most common hereditary syndromes that include gynecologic cancers as part of their cancer spectrum. Other syndromes that include gynecologic cancers include Cowden’s syndrome (PTEN germ-line mutation), Peutz-Jeghers syndrome (STK11 germline mutation), and Li-Fraumeni syndrome (TP53 germline mutation) (Table 3.1). All of these syndromes are caused by mutations in tumor-suppressor genes. In order for an individual to develop a cancer associated with one of these syndromes, both the maternally and paternally inherited copy of the relevant gene must be inactivated as described by Knudsen’s two hit hypothesis (1). Women with hereditary cancer syndromes have inherited, from either their mother or their father, a nonworking copy of one of the relevant genes and this defect is present in all of their cells. Therefore, in order for cancer to develop in a susceptible tissue through defects in a specific pathway, only the second working copy of the relevant gene needs to be lost. This is why the cancers occur earlier and more frequently than in the general population. This also explains why not all individuals with an inherited predisposition develop cancer, as it is possible that the second working copy of the relevant tumor suppressor gene is never lost in a susceptible tissue. Further, as these syndromes are all caused by mutations in a single allele of the relevant tumor suppressor gene, they are all autosomal dominant disorders in which each offspring is at 50% risk of inheriting the cancer predisposition.
Selected Hereditary Cancer Syndromes with Gynecologic Manifestations |
In recent years, there has been increasing emphasis placed on developing guidelines to assist physicians in recognizing those patients in whom genetic counseling and testing may be helpful. In addition, new prognostic and therapeutic implications that distinguish a cancer patient with a hereditary cancer syndrome have been discovered. Finally, recommendations for prevention of cancer have been refined over the last decade and will be discussed in this chapter.
HEREDITARY BREAST OVARIAN CANCER SYNDROME
Epidemiology
Of all common solid tumors, ovarian, fallopian tube, and primary peritoneal cancers have the highest proportion that is caused by heritable germline mutations, with mutations in BRCA1 and BRCA2 accounting for the vast majority. In one series from University of Washington, of 360 patients undergoing primary surgery for epithelial ovarian, fallopian tube, or primary peritoneal cancer, 63 (17.5%) patients had a deleterious mutation in either BRCA1 or BRCA2. Three (0.8%) patients had deleterious mutations in TP53 and 2 (0.5%) patients had deleterious mutations in MSH6 (2). Additionally, 17 (4.7%) patients harbored germ-line loss of function mutations in 1 of 8 other genes known to be important in inherited breast cancer (CHEK2, PALB2, BARD1, BRIP1, RAD50, RAD51C, NBN, and MRE11), though it is not yet known if risks associated with mutations in these other genes are great enough to be considered causative or if loss of function mutations in these genes act more as modifiers of cancer risk.
When just pelvic serous cancer is examined, 16% to 18% of unselected patients with high-grade pelvic serous cancer have been reported to have a deleterious BRCA1 or BRCA2 mutation (3–5). In 2005, Pal and colleagues reported on a population-based series of 121 incident pelvic serous cancers diagnosed in the Tampa, Florida region from 2000 to 2003. In this series, 20 (16.5%) patients had a deleterious BRCA mutation identified on sequencing of BRCA1 and BRCA2 (3). When the cohort was stratified by family history, mutations were found in 10 (9.9%) of 101 patients without any family history of breast or ovarian cancer in first- or second-degree relatives. In 2011, Zhang and colleagues reported an updated population-based series of 1342 invasive ovarian cancers ascertained in Ontario, Canada, from 1995 to 1999 and from 2002 to 2004. In this series, 135 (18.0%) of 751 patients with serous ovarian cancer had a deleterious BRCA1 or BRCA2 mutation identified (4). Of note, 6.1% of the nonfounder mutations identified in this study were large genomic rearrangements not detected on sequencing and only identified on multiplex ligation-dependent probe amplification (MLPA) testing. In 2012, Alsop and colleagues found 118 (16.6%) BRCA1 and BRCA2 mutations in 709 patients with incident pelvic serous cancers ascertained as part of the Australia Ovarian Cancer Study from 2002 to 2006. Additionally, similar to the Pal study, 62 (8.3%) of 749 patients without a significant family had a germline BRCA mutation identified, and 44% of all the mutations identified in the study occurred in the absence of a family history (5).
Given this substantial frequency of mutations in women with pelvic serous cancer irrespective of family history, both the American College of Obstetricians and Gynecologists and the National Comprehensive Cancer Network state that it is reasonable to offer testing for BRCA mutations to any woman with high-grade serous ovarian cancer if it will impact the care of either the woman or her close family members (6, 7).
Pathology
BRCA1– and BRCA2-associated cancers appear to be associated only with specific histological subtypes of ovarian and fallopian tube cancer. In one of the first studies to examine this issue, Boyd and colleagues genotyped 189 consecutive Jewish patients with invasive epithelial ovarian cancer, seen at Memorial Sloan-Kettering Cancer Center for the 3 common Ashkenazi founder mutations. Of the 88 patients with a founder mutation in BRCA1 or BRCA2, 60 (68%) had serous ovarian cancer and 12 (14%) had adenocarcinoma, not otherwise specified. Twelve (14%) and 2 (2%) patients had endometrioid and clear cell ovarian cancer, respectively. No patient with mucinous ovarian cancer had a BRCA mutation (8). More recently, in the 2011 Zhang report, 82% of BRCA-associated cancers were serous, 16% were endometrioid, 1.2% were clear cell, and 0% were mucinous (4).
It should be noted that, in most of the studies to date, the proportion of endometrioid and clear cell carcinomas associated with BRCA mutations may be overrepresented. BRCA-associated serous ovarian cancers have been reported to appear “pseudo-endometrioid,” resulting in misclassification unless serous specific immunohistochemical markers are used (9). In the 2012 Alsop report, BRCA1 or BRCA2 mutations were identified in 10 (8.9%) of 119 cancers initially diagnosed as endometrioid. However, 8 of these cancers were subsequently reclassified as serous or unspecified carcinoma after immunohistopathology review. Similarly, of the 4 (6.3%) of 63 cancers initially classified as clear cell identified as having a BRCA mutation, 3 were reclassified to be high-grade serous with focal clear cell differentiation (5).
Natural History of BRCA-associated Ovarian Cancer
Over a dozen studies have reported on differences in outcome between BRCA-associated and sporadic ovarian and fallopian tube cancers (8, 10–19). In the first study to address this issue, Rubin and colleagues, utilizing a case-control design, reported on 53 patients with a deleterious BRCA1 mutation. They found that patients with advanced stage BRCA1-associated disease had a median survival of 77 months compared to a median survival of 29 months in age, stage, and histologic subtype matched controls (10). In 2008, Chetrit and colleagues reported on an Israeli population-based series including data from 779 Jewish women with invasive epithelial ovarian cancer genotyped for the three Ashkenazi founder mutations. In this series, BRCA mutation status was associated with an increase in survival from 37.9 months to 53.7 months (15). Most recently, investigators from the Cancer Genome Atlas (TCGA) project reported that patients with stage II to IV high-grade serous ovarian or fallopian tube cancer and germline or sporadic mutations in BRCA1 or BRCA2 had improved outcome compared to patients without evidence of BRCA deficiency (median overall survival 66.5 vs. 41.9 months, p = 0.0003) (17). Interestingly, in this series, tumors associated with mutations in BRCA1 or BRCA2 also had improved outcome compared to patients with silencing of BRCA1 due to methylation of the BRCA1 promoter, suggesting that the mechanism of loss of BRCA function may be relevant to the biology and clinical behavior of these tumors.
Until recently, most studies examining the impact of BRCA mutations on outcome have analyzed carriers of BRCA1 mutations and carriers of BRCA2 mutations together. However, mutations in BRCA1 and BRCA2 cause related, but distinct, cancer susceptibility syndromes. Given this, it is possible that response to therapy and clinical outcome may differ between carriers of BRCA1 mutations and carriers of BRCA2 mutations. In the first study to examine this issue, Yang et al. reexamined data from the TCGA ovarian project. In this report, patients with BRCA2-associated ovarian cancer had markedly better outcome than those with BRCA wild-type tumors (HR = 0.33; 95% CI, 0.16–0.69). BRCA1-associated tumors also appeared to have a somewhat better outcome than BRCA wildtype tumors (HR = 0.76; 95% CI, 0.43–1.35), but this result did not reach statistical significance (20). In a single institution retrospective cohort from Memorial Sloan-Kettering Cancer Center, Hyman and colleagues reported on the outcome of 190 stage III to IV high-grade serous ovarian and fallopian tube cancer patients genotyped for BRCA mutations. Similar to the TCGA series, BRCA2-associated tumors were associated with significantly improved outcome compared to BRCA wildtype tumors (HR = 0.20; 95% CI, 0.06–0.65). In this study, a trend suggesting that BRCA1-associated tumors may have improved outcome compared to BRCA wildtype tumors (HR = 0.70; 95% CI, 0.36–1.38) was also noted (18). Most recently, Bolton et al. reported on a pooled series of 3,879 invasive epithelial ovarian cancers genotyped for BRCA mutations and found that in a model adjusted for age at diagnosis, stage, grade, and histology, both BRCA2 (HR = 0.49; 95% CI, 0.39–0.61) and BRCA1 (HR = 0.73; 95% CI, 0.64–0.84) had improved outcome compared to BRCA wild-type tumors. Tests of heterogeneity also demonstrated that hazard ratio for BRCA2 mutation carriers was significantly different from the HR for BRCA1 mutation carriers (19). Given the clear differences in the outcome between BRCA mutated and BRCA wildtype tumors, several authors have recommended that, at least for participants on clinical trials, we should obtain germline BRCA mutation status and stratify outcomes for the presence or absence of mutations (21, 22).
Therapeutic Implications
BRCA1 and BRCA2 are known to be necessary for repair of double strand DNA breaks through homologous recombination (23, 24). Therefore, tumor cells from BRCA-associated ovarian cancers that have no working copy of either BRCA1 or BRCA2 are believed to be exquisitely sensitive to agents that induce double strand DNA breaks, such as platinum-based chemotherapy. While this mechanism likely explains at least some of the survival advantage seen in BRCA-associated ovarian cancer, until recently knowledge of this information did not provide a new therapeutic opportunity as almost all patients with serous ovarian cancer receive platinum-based chemotherapeutics as part of first-line therapy. In 2005, however, 2 independent groups hypothesized that the BRCA-dependent homologous recombination pathway could be stressed by inhibiting poly (ADP-ribose) polymerase (PARP), an enzyme necessary for the repair of single strand DNA breaks (25, 26). If this enzyme (and repair of single strand DNA breaks) is inhibited during DNA replication, the advancing replication fork converts single stand DNA breaks into double strand breaks to be repaired by homologous recombination. In patients with a germline mutation in BRCA1 or BRCA2, the single nonmutant allele is sufficient for allowing repair in non-tumor-associated cells. However, tumor associated cells have lost both working alleles of either BRCA1 or BRCA2 and therefore cannot utilize homologous recombination, forcing the cell to use error-prone nonhomologous end joining, which frequently leads to complex rearrangements and eventual apoptosis (27).
The first clinical data supporting the potential utility of PARP inhibitors in BRCA-associated tumors were published in 2009 (28). In this phase 1 trial, 60 patients, including 22 with either a documented BRCA1 or BRCA2 mutation, received increasing doses of a novel PARP inhibitor, olaparib. Radiologic response or stable disease was observed in 11 (9 ovarian and 2 breast cancer) of 19 patients with heavily pretreated BRCA-associated ovarian, breast or prostate cancer. This was followed shortly thereafter by a phase 2 study evaluating activity of olaparib at two dose levels in 57 patients with BRCA-associated pelvic serous cancer (29). In this series, 11 of 33 patients treated at the higher dose level had complete or partial response and an additional 12 patients had stable disease. Given the promising results of these first two trials, there are over a dozen ongoing or planned trials of PARP inhibitors in BRCA-associated pelvic serous cancer (30). Further, given that 9% to 15% of sporadic ovarian cancer has silencing of the homologous recombination pathway through methylation of the BRCA1 promoter (17, 31, 32), PARP inhibitors are also being actively investigated for treatment of sporadic disease.
Genetic Counseling for Hereditary Breast and Ovarian Cancer Syndrome
Approximately 1 in 345 to 1 in 800 women have a BRCA1 or BRCA2 mutation associated with hereditary breast and ovarian cancer (33, 34). Hereditary breast and ovarian cancer syndrome is seen in all racial and ethnic groups. However, due to the phenomenon of genetic drift, several racial and ethnic groups have seen a marked increase in the population frequency of specific mutations, known as founder mutations. Best known of the founder mutations are the 185delAG and 5382insC mutations in BRCA1 and the 6174delT mutation in BRCA2 that are collectively found in 1 of 40 individuals of eastern European Jewish (Ashkenazi) heritage. Increased incidence of BRCA mutations has also been seen in individuals of Icelandic, Swedish, Dutch, Polish, French Canadian, and Hungarian descent.
The vast majority of deleterious mutations in BRCA1 and BRCA2 are nonsense mutations that lead to a premature stop codon and a truncated protein. While most protein-truncating mutations are detected by direct sequencing of BRCA1 and BRCA2, approximately 6% to 18% of clearly deleterious mutations are caused by large genomic deletions or rearrangements that will not be picked up on direct sequencing and need to instead be screened for by methods such as MLPA (35–37). Additionally 6% to 17% of individuals undergoing direct sequencing of BRCA1 or BRCA2 will have a missence mutation identified that causes a single amino acid substitution in the protein (38). These missense mutations are termed variants of uncertain significance, as it is frequently not possible to characterize whether the protein can tolerate the resulting amino acid substitution or if the substitution will cause abrogation of protein function.
Genetic counselors and other appropriately trained genetic professionals can assist in determining what are the most appropriate genetic tests for an individual patient. In addition, they can assist in interpreting the results in context with the family history. Further, the genetic counselor can assist in identifying who is the most appropriate individual to initiate genetic testing with, as in many families, it may be more informative to initiate testing in a relative other than the present individual. Lastly, genetic counselors can help identify other relatives who should be informed of a potential inherited risk (39) and can assist patients with plans for such communication.
CANCER RISKS ASSOCIATED WITH A BRCA1 OR BRCA2 MUTATION
For women with mutations in BRCA1, lifetime risks, through age 70, of pelvic (ovarian, fallopian tube, or primary peritoneal) cancer are on the order of 39% to 46%. For women with mutations in BRCA2, the lifetime risk of pelvic cancer is 12% to 20% (40, 41). The average age of diagnosis of BRCA1-associated pelvic cancers is 53, which is approximately 10 years earlier than the average age of diagnosis of sporadic ovarian or fallopian tube cancer. Interestingly, the average age of BRCA2-associated pelvic cancer is 60 to 62 years, which is no earlier than seen with sporadic disease (5, 8). When considering the timing of risk-reduction approaches, it is also important to know risks of cancer by the age of menopause. For women with BRCA1 mutations, 10% to 21% will develop pelvic cancer by age 50, but only 2% to 3% of women with BRCA2 mutations will develop pelvic cancer by the same age (41, 42).
Breast cancer risks are also markedly elevated for women with mutations in these genes, with risks of breast cancer approaching 65% to 74% by age 70 for both carriers of BRCA1 and BRCA2 mutations (40, 41). Breast cancer also occurs substantially earlier than seen in the general population, with 26% to 34% of carriers of either BRCA1 or BRCA2 mutations developing breast cancer by age 50 (40, 41, 43).
A number of genome-wide association studies (GWAS) have identified single nucleotide polymorphisms (SNPS) that modify breast and/or gynecologic cancer risk in the setting of a BRCA1 or BRCA2 mutation (44). Unfortunately, none of these markers, alone or in combination, has been shown to alter BRCA-associated cancer risk at a magnitude necessary to appreciably guide recommendations for risk-reduction strategies (45).
GUIDELINES FOR OFFERING GENETIC RISK ASSESSMENT FOR HEREDITARY BREAST AND OVARIAN CANCER SYNDROME
Several organizations have proposed guidelines for offering genetic risk assessment for hereditary breast and ovarian cancer syndrome. Likely, most relevant to the gynecologic oncologist are the guidelines jointly published by the American College of Obstetricians and Gynecologists (ACOG) and the Society of Gynecologic Oncology (SGO) (6) and those published by the National Comprehensive Cancer Network (NCCN) (7). Briefly, both of these guidelines state that hereditary cancer risk assessment is a process that: (a) should include assessment of risk, education, and counseling; (b) should be conducted by a physician, genetic counselor, or other provider with experience in cancer genetics; and (c) may include genetic testing after appropriate counseling and consent is obtained.
Specifically, ACOG and SGO recommend that women with greater than approximately a 20% to 25% chance of having a BRCA1 or BRCA2 mutation be offered a genetic risk assessment. Further, they state that it is reasonable to offer genetic risk assessment to any woman who has greater than a 5% to 10% chance of having a BRCA1 or BRCA2 mutation. Specific constellations of personal and family history that meet this threshold are outlined in Table 3.2.
There are also several risk prediction models, such as BRCAPRO, BOADICEA, and IBIS, that can assist in predicting the likelihood of a patient having a mutation in BRCA1 or BRCA2. Each of these models, however, has unique advantages and limitations, and selecting the appropriate model is generally best done with the assistance of a genetics professional.
Of particular note to physicians taking care of women with ovarian, fallopian tube, or primary peritoneal cancer, given 16% to 18% of all patients with serous ovarian cancer (including 8% to 10% of patients with no significant family history) will have a deleterious BRCA1 or BRCA2 mutation (3–5), both the AGOG/SGO and NCCN practice guidelines state that it is reasonable to offer genetic risk assessment to any women with high-grade pelvic serous cancer if it will impact either her care or the care of her close family members (6, 7).
American College of Obstetricians and Gynecologists and Society of Gynecologic Oncologists Criteria for Offering Genetic Risk Assessment for Hereditary Breast and Ovarian Cancer Syndrome |
Patients with greater than an approximate 20%–25% chance of having an inherited predisposition to breast cancer and ovarian cancer and for whom genetic risk assessment is recommended:
 Women with a personal history of both breast cancer and ovarian cancera
Women with a personal history of both breast cancer and ovarian cancera
 Women with ovarian cancera and a close relativeb with ovarian cancer or premenopausal breast cancer
Women with ovarian cancera and a close relativeb with ovarian cancer or premenopausal breast cancer
 Women with ovarian cancera who are of Ashkenazi Jewish ancestry
Women with ovarian cancera who are of Ashkenazi Jewish ancestry
 Women with breast cancer at age 50 years or younger and a close relativeb with ovarian cancera or male breast cancer at any age
Women with breast cancer at age 50 years or younger and a close relativeb with ovarian cancera or male breast cancer at any age
 Women of Ashkenazi Jewish ancestry in whom breast cancer was diagnosed at age 40 years or younger
Women of Ashkenazi Jewish ancestry in whom breast cancer was diagnosed at age 40 years or younger
 Women with a close relativeb with a known BRCA1 or BRCA2 mutation
Women with a close relativeb with a known BRCA1 or BRCA2 mutation
Patients with greater than an approximate 5%–10% chance of having an inherited predisposition to breast cancer and ovarian cancer and for whom genetic risk assessment may be helpful:
 Women with breast cancer at age 40 years or younger
Women with breast cancer at age 40 years or younger
 Women with ovarian cancer, primary peritoneal cancer, or fallopian tube cancer of high-grade, serous histology at any age
Women with ovarian cancer, primary peritoneal cancer, or fallopian tube cancer of high-grade, serous histology at any age
 Women with bilateral breast cancer (particularly if the first case of breast cancer was diagnosed at age 50 years or younger)
Women with bilateral breast cancer (particularly if the first case of breast cancer was diagnosed at age 50 years or younger)
 Women with breast cancer at age 50 years or younger and a close relativeb with breast cancer at age 50 years or younger
Women with breast cancer at age 50 years or younger and a close relativeb with breast cancer at age 50 years or younger
 Women of Ashkenazi Jewish ancestry with breast cancer at age 50 years or younger
Women of Ashkenazi Jewish ancestry with breast cancer at age 50 years or younger
 Women with breast cancer at any age and two or more close relativesb with breast cancer at any age (particularly if at least one case of breast cancer was diagnosed at age 50 years or younger)
Women with breast cancer at any age and two or more close relativesb with breast cancer at any age (particularly if at least one case of breast cancer was diagnosed at age 50 years or younger)
 Unaffected women with a close relativeb who meets one of the previous criteria
Unaffected women with a close relativeb who meets one of the previous criteria
aCancer of the peritoneum and fallopian tubes should be considered a part of the spectrum of the hereditary breast and ovarian cancer syndrome.
bClose relative is defined as a first-degree relative (mother, sister, daughter) or second-degree relative (grandmother, granddaughter, aunt, niece).
Source: From ACOG Practice Bulletin No. 103: Hereditary breast and ovarian cancer syndrome. Obstet Gynecol 2009;113:957–966, with permission
Risk-Reduction Recommendations for Carriers of BRCA1 and BRCA2 Mutations |
Breast
 Annual mammography and annual breast MRI starting by age 30
Annual mammography and annual breast MRI starting by age 30
 RRSO to reduce breast cancer risk between age 35 and 40 and when childbearing is complete
RRSO to reduce breast cancer risk between age 35 and 40 and when childbearing is complete
 Consider chemoprevention with tamoxifen, raloxifene, or an aromatase inhibitor
Consider chemoprevention with tamoxifen, raloxifene, or an aromatase inhibitor
 Consider RRM
Consider RRM
Ovary/Fallopian Tube
 RRSO to reduce ovarian and fallopian tube cancer risk between age 35 and 40 and when childbearing is complete
RRSO to reduce ovarian and fallopian tube cancer risk between age 35 and 40 and when childbearing is complete
 Consider chemoprevention with oral contraceptives
Consider chemoprevention with oral contraceptives
 Consider ovarian cancer screening with q6 month transvaginal ultrasound and CA-125 from age 30–35 years until definitive risk-reduction with RRSO
Consider ovarian cancer screening with q6 month transvaginal ultrasound and CA-125 from age 30–35 years until definitive risk-reduction with RRSO
RRSO, risk-reducing salpingo-oophorectomy; RRM, risk-reducing mastectomy.
Source: Adapted from National Comprehensive Cancer Network: NCCN Clinical Practice Guidelines in Oncology – Genetic/Familial High-Risk Assessment: Breast and Ovarian. Version 1.2012. http://www.nccn.org. 2012.
Screening and Prevention
For unaffected patients with a deleterious mutation in BRCA1 or BRCA2, there are several options to reduce the risk of both gynecologic and breast cancer (Table 3.3).
Screening
Ovarian/Fallopian Tube Cancer
Screening for ovarian and fallopian tube cancer in the general population is not recommended. Reasons for this include the limited sensitivity and specificity of transvaginal ultrasound and CA-125 serum screening. Perhaps equally important is the low annual incidence of disease. In 2008, the annual incidence of ovarian cancer in women over 50 years was 1 in 2785 per year (46). Given this incidence, in order to achieve a positive predictive value of 10% (i.e., 1 ovarian cancer detected for every 10 surgeries) a screening approach must have a specificity of 99.7% in the setting of 100% sensitivity, which has not been achievable with current technologies. However, the annual incidence of ovarian cancer in women over 50 is 1.0% to 2.5% in BRCA1 mutation carriers and 0.4% to 0.8% in BRCA2 mutation carriers (40). In this setting, assuming an annual incidence of disease of 1%, a screening regimen with 100% sensitivity only needs a specificity of 91% to achieve a positive predictive value of 10%, which is potentially achievable with current imaging and serum screening approaches. In 2002, Scheuer and colleagues reported the experience of 62 women with a documented mutation in BRCA1 or BRCA2 who participated in ovarian cancer screening with twice-yearly transvaginal ultrasound and CA-125 (47). In this cohort, 5 of 7 ovarian and peritoneal cancers were screen detected, with 4 of these cancers being stage I or stage II. During the course of this study, however, another 5 of 55 women without cancer underwent exploratory surgery due to abnormal screening results. Together these results suggest that twice-yearly transvaginal ultrasound and CA-125 screening in women with BRCA1 or BRCA2 mutations was associated with possibly acceptable 71% sensitivity, 91% specificity, and 50% positive predictive values. Importantly, a similar screening program at Cedars-Sinai Medical Center could not replicate these results. In this series of 213 women at increased risk of ovarian cancer, including 33 with mutations in BRCA1 or BRCA2, only 4 of 8 invasive ovarian cancers were screen detected, and 3 of the 4 screen detected cancers were stage IIIc (48). In 2007, Hermsen and colleagues reported on the experience of 888 women with BRCA1 or BRCA2 mutations who participated in ovarian cancer screening with annual transvaginal ultrasound and CA-125 for a mean of 1.7 years between 1993 and 2005. In this study, 5 of the 10 incident invasive cancers presented as interval cancers with a normal screening result 3 to 10 months prior to diagnosis. Further, of the 5 screen detected cancers, 4 were stage III or IV, leading the authors to conclude that annual screening is not efficacious (49).
Of note, all of the studies to date commenting on ovarian cancer screening in BRCA mutation carriers have been retrospective case series and not rigorously conducted prospective screening trials. Two such prospective trials specifically targeting women at familial/inherited risk are either ongoing or recently completed, but not reported. In the recently completed Gynecologic Oncology Group study 0199, a prospective study of risk-reducing salpingo-oophorectomy and longitudinal CA-125 screening among women at increased genetic risk of ovarian cancer, participants elected either surveillance with annual transvaginal ultrasound and every 3 month CA-125 interrogated with the risk of ovarian cancer (ROCA) algorithm or risk-reducing salpingo-oophorectomy (RRSO) (50). All participants in the study were genotyped, and just under 400 of the approximately 1600 women who elected surveillance had a BRCA mutation. The study has completed data collection and is currently in analysis. The UK familial ovarian cancer screening study (UK FOCSS) is the other ongoing large-scale trial of women at familial or inherited risk (51). In this study, approximately 2350 women with greater than a 10% lifetime risk of ovarian cancer are being followed with annual transvaginal ultrasound and every 4 month CA-125 also interrogated with the ROCA algorithm. Study results are expected in 2013–2014.
Until results from GOG 199 and UK FOCSS are available, despite the absence of clear evidence for benefit, women with mutations in BRCA1 or BRCA2 likely should be recommended to consider ovarian cancer screening with transvaginal ultrasound and CA-125 determinations every 6 months from age 30 to 35 until they undergo definitive risk-reduction with risk-reducing salpingo-oophorectomy (7).
Breast Cancer
For patients with a deleterious mutation in BRCA1 or BRCA2, annual mammography starting by age 30 is recommended (7, 52). However, several studies have suggested that mammography alone in women with BRCA mutations is inadequate. In 2001, Brekelmans and colleagues reported that 4 of 9 invasive breast cancers in 128 BRCA mutation carriers presented as palpable masses in the interval between screens (53). Similarly, in 2002, Scheuer et al. reported that 7 of 12 invasive breast cancers diagnosed in 251 BRCA mutation carriers undergoing mammographic screening presented as interval cancers (47). Due to the relatively low sensitivity of mammography, breast MRI has been investigated as a complimentary imaging modality. Three of the largest studies to date have shown a 71% to 78% sensitivity of breast MRI for BRCA-associated cancers compared to a 36% to 40% sensitivity for mammography (54). Further, in 2011, Warner and colleagues demonstrated that the addition of annual MRI to annual mammography led to a down staging of BRCA-associated breast cancers (adjusted hazard ratio for the development of stage II to IV breast cancer associated with MRI screening = 0.30; 95% CI, 0.12–0.72) (55). Currently both the National Comprehensive Cancer Network and the American Cancer Society recommend the combination of annual mammography and annual breast MRI, beginning by age 30, for breast screening in women with BRCA mutations (7, 52).
Chemoprevention
Ovarian/Fallopian Tube Cancer
Multiple studies have suggested that use of oral contraceptives in the general population is associated with a substantial and long-lasting reduction in the risk of ovarian cancer (56). Given this, several authors have examined the impact of oral contraceptives on ovarian cancer risk in the setting of BRCA1 or BRCA2 mutations. In a recent meta-analysis of 5 case-control and retrospective cohort studies, Iodice and colleague found that ever use of oral contraceptives was associated with a significant reduction in ovarian cancer risk in both BRCA1 (summary relative risk = 0.51; 95% CI, 0.40–0.65) and BRCA2 (summary relative risk = 0.52; 95% CI, 0.31–0.87) mutation carriers (57). Further, longer use was associated with a greater risk-reduction, with a 36% risk reduction seen for each 10 years of use (summary relative risk = 0.64; 95% CI, 0.53–0.78).
When caring for women with BRCA1 or BRCA2 mutations, it is important, however, to evaluate for both breast and ovarian cancer risk, and the data regarding breast cancer risk has been somewhat more conflicting. Of 5 published studies addressing the impact of oral contraceptives on breast cancer risk in BRCA1 mutation carriers, 3 suggested an increased risk of breast cancer with oral contraceptive use and 2 showed no increase in risk (58–62). Similarly in the 3 studies reporting on breast cancer risk with oral contraceptive use in BRCA2 mutation carries, 2 studies suggested an increased risk, and one study showed no increase in risk (58, 60, 62). In the Iodice meta-analysis, neither BRCA1 (summary relative risk = 1.09; 95% CI, 0.77–1.54) nor BRCA2 (summary relative risk = 1.15; 95% CI, 0.61–2.18) mutation carriers demonstrated a statistically significant increased risk of breast cancer with oral contraceptive use. However, oral contraceptive formulations used before 1975 were associated with an increased risk of breast cancer (summary relative risk = 1.47; 95% CI, 1.06–2.04). Given the data currently available, it likely still makes sense to counsel patients that oral contraceptives may be associated with some adverse impact on breast cancer risk. This potential risk, however, needs to be balanced against the risk of unintended pregnancy and the benefit of oral contraceptives on ovarian cancer risk.
Breast Cancer
The selective estrogen receptor modulators (SERMs), tamoxifen and raloxifene, in addition to the aromatase inhibitor exemestane have been studied in the general population as chemoprevention for breast cancer. Tamoxifen is the only one of these agents that has been studied in women with BRCA mutations. King and colleagues, in a reanalysis of the NSABP P-1 trial, found a suggestion that tamoxifen was associated with protection against BRCA2 associated breast cancer (risk ratio, 0.38; 95% CI, 0.06–1.56) but not BRCA1 associated breast cancer (risk ratio, 1.67; 95% CI, 0.32–10.70), though neither of these results reached statistical significance (63). It was speculated that the reason for this possible differential effect was the different estrogen receptor phenotypes of breast cancer seen between BRCA1 and BRCA2 mutations carriers. BRCA2-associated breast cancers are ER positive 65% to 80% of the time, as opposed to only 10% to 25% of BRCA1-associated breast cancers (64, 65). In 2000, Narod and colleagues looked at the impact of therapeutic tamoxifen on contralateral breast cancer risk. In this study, tamoxifen appeared to be associated with a reduction of contralateral breast cancer risk in both BRCA1 (odds ratio 0.38; 95%; CI, 0.19–0.74) and BRCA2 (odds ratio 0.63; 95% CI, 0.20–1.50) mutation carriers (66). Of potential significance, BRCA1 mutation carriers who received tamoxifen in this study likely had ER positive disease. Weitzel et al. have subsequently demonstrated that, in women with BRCA mutations who develop contralateral breast cancer, the ER status of the second breast cancer is highly concordant with the ER status of the first breast cancer, suggesting that women with ER positive BRCA1-associated breast cancer are more likely to develop a second ER positive cancer and therefore more likely to benefit from hormonal chemoprevention (67).
Risk-Reducing Surgery
Salpingo-Oophorectomy
Impact on ovarian cancer risk. Two of the earliest studies to examine the impact of risk-reducing salpingo-oophorectomy (RRSO) on ovarian cancer risk were published in 2002. The first of these was a prospective cohort study from Memorial Sloan-Kettering Cancer Center. In this study, 170 women with a documented deleterious mutation in BRCA1 or BRCA2 elected either surveillance or RRSO. In this series, RRSO was associated with a 75% reduction in the subsequent risk of breast or BRCA-associated gynecologic cancer (HR = 0.25; 95% CI, 0.08–0.74) (68). When the impact of RRSO on gynecologic risk alone was examined, there was a suggestion of an approximately 85% reduction in gynecologic cancer risk (HR = 0.15; 95% CI, 0.02–1.31). Simultaneous with this report, a retrospective study from University of Pennsylvania found that RRSO was associated with an approximately 96% reduction in gynecologic cancer risk (HR = 0.04; 95% CI, 0.01–0.16) (69). However, a commentary from Klaren et al. pointed out that the retrospective study may have over-estimated the risk-reduction conferred (70). Since that time, several other studies have suggested that oophorectomy is associated with an 71% to 89% reduction in gynecologic cancer risk and a 2009 meta-analysis concluded that RRSO was associated with approximately an 79% reduction in risk of BRCA-associated gynecologic cancer (HR = 0.21; 95% CI, 0.12–0.39) (71).
The origin of peritoneal cancers after RRSO is not entirely clear. Some of these may represent recurrence of occult ovarian or tubal malignancies that were not recognized at time of initial pathologic evaluation, emphasizing the need for careful pathologic evaluation of the entire ovary and fallopian tube at time of RRSO. It has also been speculated that the peritoneal cancer can arise from exfoliated tubal cells (endosalpingiosis) that implant on the peritoneum and undergo malignant transformation in that location. Lastly, some authors have suggested that peritoneal malignancies can start exclusively in the peritoneum through mullerian metaplasia (72). Irrespective of the origin, it is important that patients undergoing RRSO be informed of the small possibility of primary peritoneal cancer occurring after the procedure.
Impact on breast cancer risk. In the first study examining the impact of RRSO on breast cancer risk, Rebbeck and colleagues found that RRSO in women with a BRCA1 mutation was associated with a 47% reduction in BRCA1-associated breast cancer risk (HR = 0.53; 95% CI, 0.33–0.84) (73). In the prospective study by Kauff et al. reported in 2002, which examined both BRCA1 and BRCA2 mutation carriers, RRSO appeared to be associated with approximately a 68% reduction in breast cancer risk (HR = 0.32; 95% CI, 0.08–1.20) (68). In 2008, Kauff and Rebbeck pooled their updated prospective data and were able to report on 597 women with breast tissue at risk who had RRSO or surveillance and were prospectively followed for 2.8 years (74). When women with BRCA1 and BRCA2 mutations were examined together, RRSO was associated with a 47% reduction in breast cancer risk (HR = 0.53; 95% CI, 0.29–0.96). However when BRCA1 and BRCA2 mutation carriers were examined separately, women with BRCA2 mutations had a 72% reduction in breast cancer risk compared to women who left their ovaries in situ (HR = 0.28; 95% CI, 0.08–0.92). Women with BRCA1 mutations appeared to have a 39% reduction in breast cancer risk (HR = 0.61; 95% CI, 0.30–1.22), but despite this being the largest prospective study to date, these results did not reach statistical significance. An exploratory analysis examining the impact of RRSO on breast cancer stratified for ER status was also performed. In this analysis, RRSO appeared to be profoundly protective against ER positive breast cancer (HR = 0.22; 95% CI, 0.05–1.05). Not unexpectedly, no impact against ER-negative breast cancer was noted (HR = 1.10; 95% CI, 0.48–2.51).
Impact on life expectancy. Domchek and colleagues reported on results of a prospective cohort study concluding that RRSO was associated with reduction in breast cancer–specific (HR = 0.44; 95% CI, 0.26–0.76), ovarian cancer–specific (HR = 0.21; 95% CI, 0.06–0.80), and all-cause mortality (HR = 0.40; 95% CI, 0.26–0.61) (75). While the effect of RRSO on mortality demonstrated by Domchek and colleagues is almost certainly present, there is a fair amount of instability in the estimates of the actual magnitude of this effect, as participants could be ascertained to this study as many as 20 years prior to the identification of BRCA1 and BRCA2, but in many cases likely only underwent genotyping (and inclusion in the final analysis) if: (a) they lived long enough for clinical testing to become available and (b) they developed a cancer of interest.
Even given the limitations of this and other studies evaluating the efficacy of RRSO on subsequent cancer risk and mortality, for carriers of BRCA1 and BRCA2 mutations, RRSO remains the best available protection against pelvic serous cancer and should be strongly considered between 35 and 40 years of age and after childbearing is complete.
Technical considerations. Given that the ovaries and fallopian tubes are both at risk for malignant transformation, it is imperative that all of the ovaries and distal fallopian tube are removed at time of RRSO. In order to do this, it is necessary that the surgeon be able to enter the retroperitoneal space and ligate the infundibulopelvic ligament at least 2 cm from its insertion into the ovary.
Malignant cells leading to upstaging of disease have also been found in peritoneal cytology specimens from a number of women undergoing RRSO (76). Given these findings, washing likely should be performed at time of peritoneal entry in all women undergoing RRSO.
Additionally as 2% to 10% of RRSO specimens obtained from BRCA mutation carriers have an occult malignancy detected at time of pathologic review (77), it is essential that the entire ovary and fallopian tube be serially sectioned to minimize the possibility of a small invasive cancer going undetected (6). The fimbrial ends of the fallopian tubes are the most frequent site of occult invasive serous carcinomas (78–80), and these lesions are best visualized using the SEE-FIM (sectioning and extensively examining the fimbriated end) method (81). Briefly, in this method, the fimbriated portion of the fallopian tube is sectioned lengthwise (sagittally) in multiple planes to maximize exposure of the tubal plicae. When this method is utilized, serous tubal intraepithelial carcinoma (STIC), a putative precursor to invasive serous carcinoma, is also identified in as many as 5% to 8% of specimens obtained at time of RRSO in women with mutations in BRCA1 or BRCA2 (82, 83). Both the occult fallopian tube malignancies and STIC lesions typically overexpress mutant TP53 protein (84). Patients in whom small high-grade invasive serous cancers are found incidentally at RRSO generally receive adjuvant chemotherapy. The role of chemotherapy in the management of serous tubal intraepithelial carcinoma (STIC) is less clear (76).
Timing of procedure. For most women with mutations in either BRCA1 or BRCA2, RRSO should generally be considered between age 35 and 40 and when childbearing is complete (6). For women with BRCA1 mutations, this age is recommended because only 2% to 3% of women with mutations in this gene will develop pelvic serous cancer by age 40, but 10% to 21% of BRCA1 mutation carriers will develop pelvic serous cancer by age 50 years (40–42). For women with BRCA2 mutations, the risk of pelvic serous cancer by age 50 is only 2% to 3%. However, women with BRCA2 mutations who defer RRSO until the age of natural menopause, likely lose the profound reduction against BRCA2-associated breast cancer conferred by RRSO (74). For women with BRCA2 mutations who have already had bilateral mastectomy and ovarian ablation is not being utilized as part of adjuvant therapy for a prior breast cancer, RRSO likely can be reasonably deferred until the mid-40s.
Mastectomy
Several studies have demonstrated that risk-reducing mastectomy (RRM) in women with BRCA1 or BRCA2 mutation is associated with an approximately 90% reduction in the risk of new breast cancer (85, 86). Importantly, the impact on life expectancy may be markedly less, as the majority of these cancers in women undergoing both mammography and breast MRI will be diagnosed at a curable stage. In a decision analysis by Kurian et al. RRM at age 40 (in addition to RRSO at age 40 and breast screening starting at age 25) only increased the probability of survival to age 70 from 74% to 77% in carriers of BRCA1 mutations and from 80% to 82% in carriers of BRCA2 mutations (87). Given these relatively small absolute changes in survival, either intensive breast screening or risk-reducing mastectomy is likely a reasonable option for a woman with a BRCA1 or BRCA2 mutation.
LYNCH SYNDROME
Epidemiology
Approximately 5% of endometrial cancer cases may be attributed to an inherited predisposition (88). Lynch syndrome, or hereditary nonpolyposis colorectal cancer (HNPCC) syndrome, accounts for the majority of these cases. Individuals with Lynch syndrome have a germline mutation in one of four genes in the DNA mismatch repair family; MLH1, MSH2, MSH6, or PMS2. While Lynch syndrome has historically been characterized by an increased risk for colorectal cancer, women with Lynch syndrome also have a substantial risk for endometrial cancer. The estimated risk for colon cancer in women is 40% to 60% and in men as high as 80% (89, 90). In women, the lifetime endometrial cancer risk is approximately 40% to 60%. In a study specifically looking at individuals with MSH6 mutations, risk for endometrial cancer was 26% by age 70 and as high as 44% by 80 years of age, and lifetime risk of colon cancer was 10% by age 70 and 20% by age 80 (91). Women with Lynch syndrome also have an approximate 5% to 10% lifetime risk for developing ovarian cancer. Other cancers associated with Lynch syndrome include cancers of the stomach, small bowel, renal pelvis and ureter, and brain.
In the general population, Lynch syndrome occurs in about 1 in 600 to 1 in 3000 individuals (92, 93). In a population-based study on endometrial cancer patients, the incidence of Lynch syndrome was 2.3%, which is similar to the 2.2% incidence of Lynch syndrome among colon cancer patients (94). For women with endometrial cancer under the age of 50, the proportion with Lynch syndrome increases to 5% to 9% (95–97). The mean age of diagnosis for endometrial cancer in women with Lynch syndrome is 47 years, which is substantially lower than the mean age of diagnosis of endometrial cancer in the general population, 60 years. Women with Lynch syndrome are at increased risk of developing synchronous or metachronous cancers. In a study that examined 101 women with Lynch syndrome who had developed both GI cancer and gynecologic cancer, 51% presented, at a median age of 44, with their gynecologic cancer first, and 49% presented with their GI cancer first (98).
Prior to the discovery of the genes responsible for Lynch syndrome, criteria were formulated to identify families with Lynch syndrome for research studies. The initial Amsterdam I criteria focused on colon cancer and were subsequently revised (Amsterdam II), to include all Lynch syndrome–associated cancers. The criteria include: (a) three or more relatives with Lynch syndrome–associated cancers, (b) two affected relatives in successive generations, (c) one affected relative is a first-degree relative of the other two, and (d) one or more relatives with Lynch syndrome–associated cancer diagnosed before the age of 50 years.
Pathology
Unlike the ovarian cancers associated with BRCA1 and BRCA2 mutations, the endometrial cancers associated with Lynch syndrome span a broader spectrum. In a study by Broaddus et al., 43 of 50 (86%) endometrial cancers were endometrioid histology, with the remainder being papillary serous carcinoma, clear cell carcinoma, and malignant mixed mullerian tumors (99). Interestingly, all of the nonendometrioid tumors in this study occurred in patients with MSH2 mutations. Among all of the patients with Lynch syndrome, 78% were diagnosed at stage I, 10% at stage II, and 12% at stage III or IV. Lymph-vascular space involvement was noted in 24% of the cases, and 26% had deep myometrial involvement that was defined as invasion greater than 50%. Endometrial cancers that arise in the lower uterine segment are rare in the general population, but are seen more frequently in women with Lynch syndrome. In a study by Westin et al., almost one third of patients with endometrial cancers arising in the lower uterine segment were suspected to have Lynch syndrome, based on tumor studies or germline testing (100). This characteristic phenotype is similar to the increased proportion of right-sided colon cancers seen in individuals with Lynch syndrome. Other pathologic features that have been seen more frequently in Lynch syndrome–associated colon cancers, including poor tumor differentiation and tumor infiltrating lymphocytes, have not been seen consistently in Lynch syndrome associated endometrial cancers.
The ovarian cancers seen in Lynch syndrome also have a different stage and histology spectrum from that seen in women with sporadic disease. In a recent series, 22 (47%) of 47 Lynch-associated epithelial ovarian cancers presented at stage I. Further, serous cancers were underrepresented in this series, accounting for only 28% of tumors. Endometrioid, clear cell, and mucinous histologies were seen in 35%, 17%, and 5%, respectively (101).
Genetic Risk Assessment for Lynch Syndrome
For gynecologic oncologists, identifying Lynch syndrome in a patient with endometrial cancer has important implications for both the woman with cancer and for her family members. Individuals with Lynch syndrome are at significant lifetime risk of developing a second primary malignancy. Therefore, when an endometrial cancer patient is identified as having Lynch syndrome, appropriate colon cancer screening can be initiated. In addition, if a specific mutation in one of the DNA mismatch repair genes is identified in the endometrial cancer patient, her family members can undergo targeted predictive genetic testing for the same mutation.
Historically, guidelines to assist physicians in identifying patients with Lynch syndrome have focused on colon cancer patients. The Bethesda criteria, which were established in 1997 and revised in 2004, assist physicians in identifying colon cancer patients who may have Lynch syndrome (102). However, it has become increasingly clear that Lynch syndrome patients often present with endometrial or ovarian cancer as their first cancer. In 2007, the Society of Gynecologic Oncologists provided guidelines to assist in identifying patients for whom genetic risk assessment may be helpful (Table 3.4). They specifically recommended that women who have a greater than approximately 20% to 25% chance of having Lynch syndrome be offered risk assessment (103). Individuals meeting this criteria include women with endometrial cancer who fulfill the Amsterdam criteria; patients with synchronous or metachronous endometrial and colorectal cancer whose first cancer was diagnosed at less than age 50, patients with synchronous or metachronous ovarian and colorectal cancer whose first cancer diagnosed at less than 50, patients with endometrial cancer with molecular or immunohistochemical evidence of a mismatch repair defect, and patients with a first or second degree relative with a known mismatch repair gene mutation. In addition, the Society of Gynecologic Oncologists guidelines also identified a category of patients who have a 5% to 10% risk for Lynch syndrome and for whom genetic risk assessment may be helpful. These include patients with endometrial cancer diagnosed at less than age 50, patients with an endometrial or ovarian cancer and a synchronous or metachronous colon cancer at any age, and patients with endometrial cancer diagnosed at any age with 2 or more first-or second-degree relatives with Lynch syndrome–associated cancers, regardless of age.
Society of Gynecologic Oncologists Criteria for Offering Genetic Risk Assessment for Lynch Syndrome |
Patients with greater than approximately 20%–25% chance of having Lynch syndrome and for whom genetic risk assessment is recommended:
 Patients with endometrial or colorectal cancer who meet the revised Amsterdam criteria as listed below:
Patients with endometrial or colorectal cancer who meet the revised Amsterdam criteria as listed below:
 At least three relatives with a sentinel Lynch-associated cancer (colorectal cancer, cancer of the endometrium, small bowel, ureter, or renal pelvis) in one lineage;
At least three relatives with a sentinel Lynch-associated cancer (colorectal cancer, cancer of the endometrium, small bowel, ureter, or renal pelvis) in one lineage;
 One affected individual should be a first-degree relative of the other two;
One affected individual should be a first-degree relative of the other two;
 At least 2 successive generations should be affected;
At least 2 successive generations should be affected;
 At least 1 Lynch-associated cancer should be diagnosed before age 50.
At least 1 Lynch-associated cancer should be diagnosed before age 50.
 Patients with synchronous or metachronous endometrial and colorectal cancer with the first cancer diagnosed prior to age 50
Patients with synchronous or metachronous endometrial and colorectal cancer with the first cancer diagnosed prior to age 50
 Patients with synchronous or metachronous ovarian and colorectal cancer with the first cancer diagnosed prior to age 50
Patients with synchronous or metachronous ovarian and colorectal cancer with the first cancer diagnosed prior to age 50
 Patients with colorectal or endometrial cancer with evidence of a mismatch repair defect (i.e., microsatellite instability (MSI) or immunohistochemical loss of expression of MLH1, MSH2, MSH6, or PMS2)
Patients with colorectal or endometrial cancer with evidence of a mismatch repair defect (i.e., microsatellite instability (MSI) or immunohistochemical loss of expression of MLH1, MSH2, MSH6, or PMS2)
 Patients with a first-or second-degree relative with a known mismatch repair gene mutation
Patients with a first-or second-degree relative with a known mismatch repair gene mutation
Patients with greater than approximately 5%–10% chance of having Lynch syndrome and for whom genetic risk assessment may be helpful:
 Patients with endometrial or colorectal cancer diagnosed prior to age 50
Patients with endometrial or colorectal cancer diagnosed prior to age 50
 Patient with endometrial or ovarian cancer with a synchronous or metachronous colon or other Lynch syndrome–associated tumora at any age
Patient with endometrial or ovarian cancer with a synchronous or metachronous colon or other Lynch syndrome–associated tumora at any age
 Patients with endometrial or colorectal cancer and a first-degree relative with a Lynch syndrome–associated tumora diagnosed prior to age 50
Patients with endometrial or colorectal cancer and a first-degree relative with a Lynch syndrome–associated tumora diagnosed prior to age 50
 Patients with colorectal or endometrial cancer diagnosed at any age with two or more first of second degree-relativesb with Lynch syndrome–associated tumorsa, regardless of age
Patients with colorectal or endometrial cancer diagnosed at any age with two or more first of second degree-relativesb with Lynch syndrome–associated tumorsa, regardless of age
 Patients with a first or second-degree relativeb that meets the above criteria
Patients with a first or second-degree relativeb that meets the above criteria
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


