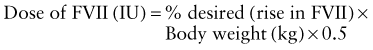Chapter 470 Hereditary Clotting Factor Deficiencies (Bleeding Disorders)
Hemophilia A (factor VIII deficiency) and hemophilia B (factor IX deficiency) are the most common and serious congenital coagulation factor deficiencies. The clinical findings in hemophilia A and hemophilia B are virtually identical. Hemophilia C is the bleeding disorder associated with reduced levels of factor XI (Chapter 470.2). Reduced levels of the contact factors (factor XII, high molecular weight kininogen, and prekallikrein) are associated with significant prolongation of activated partial thromboplastin time (APTT; also referred to as PTT), but are not associated with hemorrhage, as discussed in Chapter 470.3. Other coagulation factor deficiencies that are less common are briefly discussed in subsequent subchapters.
470.1 Factor VIII or Factor IX Deficiency (Hemophilia A or B)
Pathophysiology
Factors VIII and IX participate in a complex required for the activation of factor X. Together with phospholipid and calcium, they form the “tenase,” or factor X–activating, complex. Figure 469-1 shows the clotting process as it occurs in the test tube, with factor X being activated by either the complex of factors VIII and IX or the complex of tissue factor and factor VII. In vivo, the complex of factor VIIa and tissue factor activates factor IX to initiate clotting. In the laboratory, prothrombin time (PT) measures the activation of factor X by factor VII and is therefore normal in patients with factor VIII or factor IX deficiency.
Clinical Manifestations
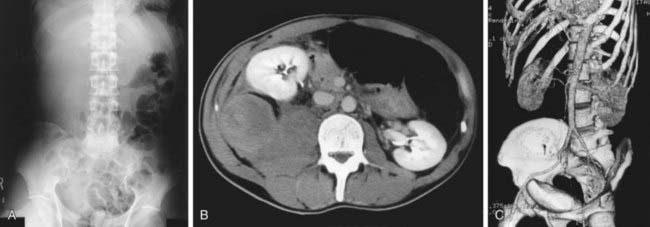
Although most muscular hemorrhages are clinically evident owing to localized pain or swelling, bleeding into the iliopsoas muscle requires specific mention. A patient may lose large volumes of blood into the iliopsoas muscle, verging on hypovolemic shock, with only a vague area of referred pain in the groin. The hip is held in a flexed, internally rotated position owing to irritation of the iliopsoas. The diagnosis is made clinically from the inability to extend the hip but must be confirmed with ultrasonography or CT (Fig. 470-1). Life-threatening bleeding in the patient with hemophilia is caused by bleeding into vital structures (central nervous system, upper airway) or by exsanguination (external trauma, gastrointestinal or iliopsoas hemorrhage). Prompt treatment with clotting factor concentrate for these life-threatening hemorrhages is imperative. If head trauma is of sufficient concern to suggest radiologic evaluation, factor replacement should precede radiologic evaluation. Simply put: “Treat first, image second!” Life-threatening hemorrhages require replacement therapy to achieve a level equal to that of normal plasma (100 IU/dL, or 100%).