Deirdre Kelly, Nicola Ruth • Understand the physiological basis of normal liver function, including absorption and secretion • Know the genetic and environmental factors in the aetiology of liver disease • Understand the pathophysiology of infective agents in the liver • Understand the pharmacological basis of therapy in liver disorders • Know the possible impact on the hepatobiliary system of other system disorders and vice versa
Hepatology
The aetiology of liver disease varies according to age. In neonates, infective, genetic and metabolic causes are the most common. As the child gets older, autoimmune/infective causes predominate.
Embryology of the hepatobiliary system
The liver is the largest intra-abdominal organ, with synthetic, metabolic, exocrine and endocrine functions. Liver development occurs through a progressive series of reciprocal tissue interactions between the embryonic endoderm and mesoderm. Application of this knowledge has allowed production of ‘hepatic-like’ tissue from embryonic stem cells, which may ultimately lead to their use in transplantation.
Overview of liver development
During gastrulation, a process which takes place in all animals, where the single layered blastula becomes a tri-layered gastrula comprising endoderm, ectoderm and mesoderm, the endoderm layer forms and leads to a primitive gut tubular structure comprising foregut, midgut and hindgut regions (Fig. 21.1).
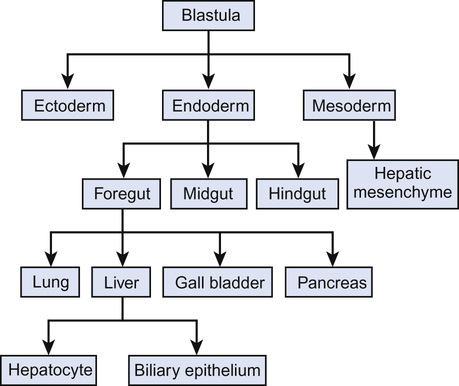
The liver originates from the ventral foregut. The first sign is formation of the hepatic diverticulum adjacent to the developing heart (Fig. 21.2). Anteriorly it becomes the liver and intrahepatic biliary tree. Posteriorly it forms the gall bladder and extrahepatic bile ducts. Hepatoblasts invade the septum transversum, forming the liver bud. This undergoes accelerated growth as it is vascularized and colonized by haematopoietic cells, becoming the major fetal haematopoietic organ. Hepatoblasts are bi-potential, becoming either the lumen of the intrahepatic bile ducts or differentiating into hepatocytes. Between antenatal day 17 and into the perinatal period, focal dilations appear forming intrahepatic bile ducts, while the remainder regress.
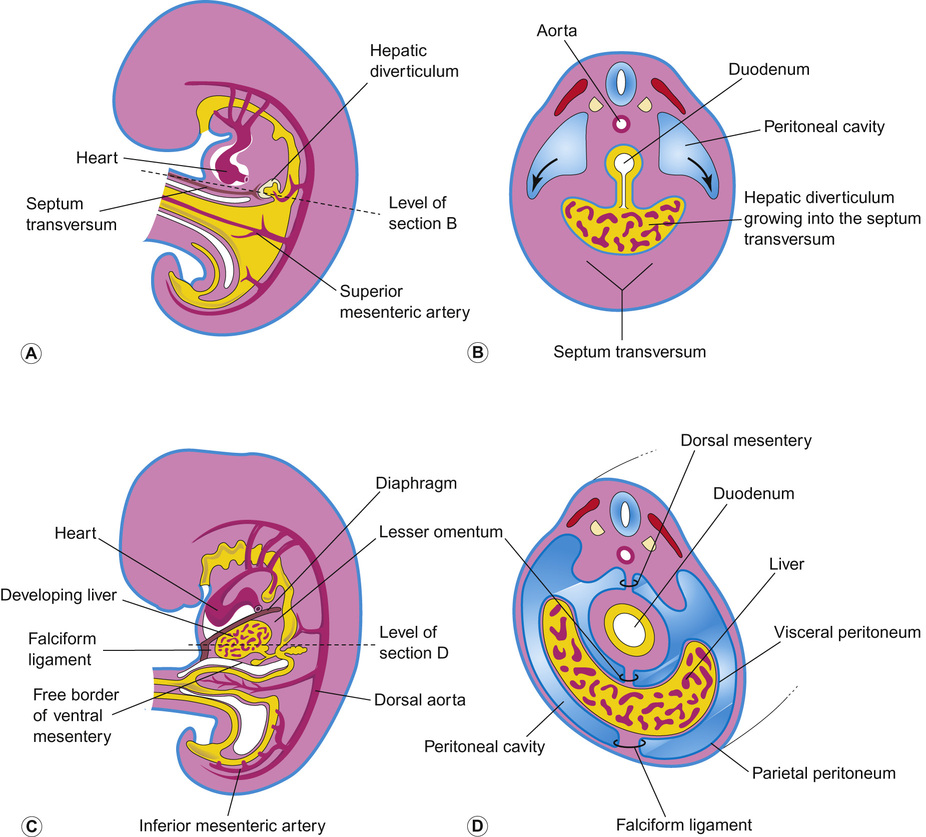
Defects in early liver bud growth can be lethal. The molecular genetics of Alagille syndrome exemplify the interaction between endoderm and mesenchyme (see also Table 21.2). Mutations in the Notch ligand gene Jagged-1 leads to a paucity of intrahepatic bile ducts. Jagged-1 expressed in the portal mesenchyme activates NOTCH-2 in adjacent hepatoblast, required to maintain bile duct morphogenesis.
Anatomy of the liver and biliary tree
The falciform ligament divides the liver anteriorly; the round ligament and umbilical fissure inferiorly. The right lobe is further divided by the gall bladder fossa into the right hemiliver and quadrate lobe. The fourth lobe (caudate) is posterior and surrounds the inferior vena cava. The biliary tree connects the liver and duodenum, the primary purpose being bile transport and storage, under neuronal and hormonal regulation. Bile is formed in hepatocytes, and is transported into the extrahepatic ducts via the cannaliculi. Bile flows into the small intestine or into the cystic duct and then into the gall bladder, regulated by the sphincter of Oddi (Fig. 21.3).
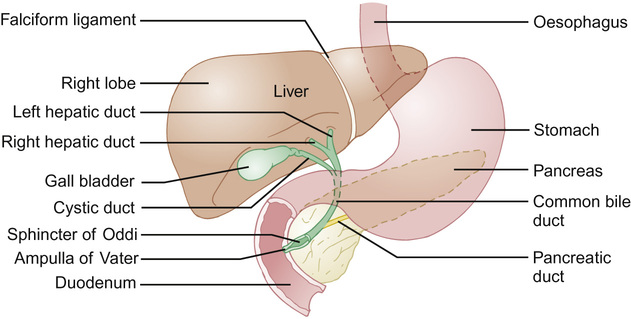
Blood supply and venous drainage
The arterial supply to the liver in utero is the left hepatic artery derived from the left gastric artery; the middle hepatic artery (common hepatic artery) derived from the coeliac trunk; and the right hepatic artery derived from the superior mesenteric artery. The blood supply assumes the adult pattern during early infancy, with atrophy of right and left hepatic arteries.
Physiology of the biliary tract
Gut bile allows absorption of fat and excretion of cholesterol, bilirubin, iron and copper. Bile is secreted by the hepatocytes into the canalicular space by active and passive processes. It is the active process which generates bile flow. The products of active secretion comprise conjugated bile acids, conjugated bilirubin, glutathione, conjugates of steroid hormones and leukotrienes. Filterable solutes (plasma, glucose, electrolytes, low-molecular-weight organic acids and calcium) are generated by passive secretion induced by osmotic pressure and are called secondary solutes.
Between meals, the gall bladder relaxes and the sphincter of Oddi contracts, leading to the diversion of hepatic bile into the gall bladder for storage until the next meal (this observation becomes important when assessing a child with conjugated jaundice in whom you suspect biliary atresia, as a fasted ultrasound scan will fail to demonstrate a gall bladder). During a meal, the gall bladder contracts, the sphincter of Oddi relaxes and bile enters the duodenum. Bile acids are absorbed from the terminal ileum and return to the liver via the portal system. This is a process of both passive and active reabsorption. The most important mechanism is a sodium-coupled transporter present in the apical membrane of the enterocytes; the ileal bile acid transporter. In the distal ileum and large intestine, intestinal bacteria deconjugate bile acids, which are absorbed passively. A small amount of the bile acid is lost in the faeces.
Function of the liver and its impact on homeostasis
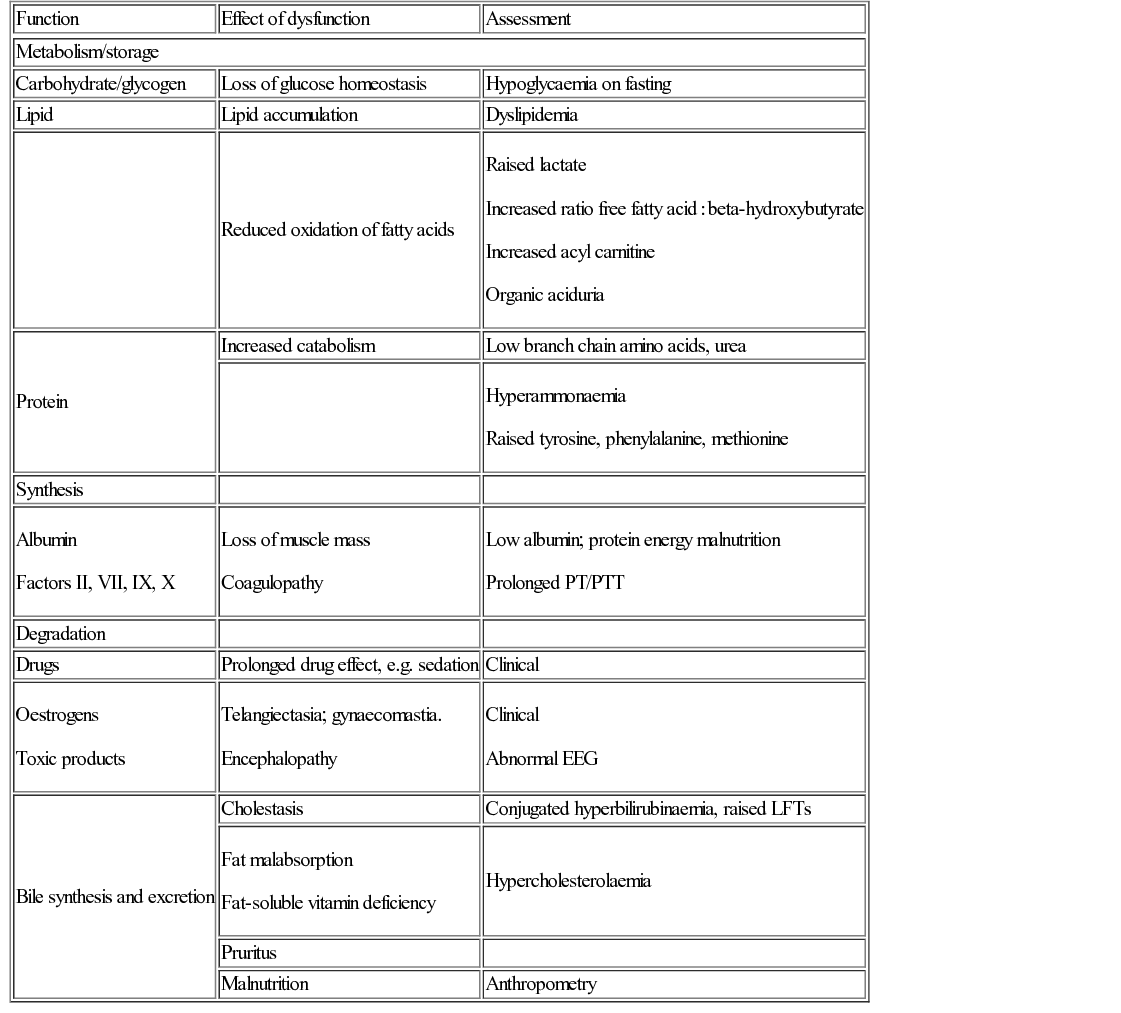
In order to understand liver disease, it is necessary to understand how the liver regulates a number of important processes (Table 21.1).
Biochemical tests
Bilirubin, in particular conjugated (direct) bilirubin, is almost always raised in liver disease irrespective of its cause. It is produced from the breakdown of red blood cells, making unconjugated (indirect) bilirubin, which circulates bound to albumin although some is ‘free’ and hence able to enter the brain. Unconjugated bilirubin is metabolized in the liver to produce conjugated bilirubin which passes into the gut and is excreted in stool.
Transaminases are present in the liver, heart and skeletal muscle. They indicate the nature of liver dysfunction when taken in combination with a background history, e.g. raised alanine aminotransferase (ALT) and aspartate aminotransferase (AST) indicate hepatic necrosis and are indicators of hepatic damage. Alkaline phosphatase, although raised in liver disease, particularly biliary disease, can also be raised in rapid bone growth or secondary to vitamin D deficiency. Gamma-glutamyl transferase (GGT) is particularly associated with biliary obstruction and inflammation.
Assessment of liver synthetic function is useful when assessing liver disease. Albumin and coagulation are sensitive markers of liver dysfunction. Hypoglycaemia is a common finding in liver dysfunction.
Radiology
Radiological investigations such as an ultrasound scan can give valuable information about the liver’s architecture, such as the shrunken cirrhotic liver with heterogeneous appearance often seen in chronic liver disease, or the enlarged liver seen in some metabolic diseases such as glycogen storage disease type I. Developmental defects can be seen on fasting ultrasound scan, such as extrahepatic biliary atresia, where a gall bladder is not demonstrable after a fast (this is abnormal and must always be discussed with a tertiary centre). The other developmental defect commonly seen on ultrasound scan is a choledochal cyst (an outpouching of the bile ducts). Surgical correction is necessary as these can result in malignancy. Gallstones can be easily demonstrated on ultrasound scan as well as any ductal dilatation. It is also possible to assess the liver’s blood supply, valuable in children who have liver disease or who have had a liver transplant, as vessel thrombosis or reversed vessel flow requires immediate assessment and management and is a poor prognostic sign. Other abdominal structures can be assessed, e.g. splenic enlargement associated with portal hypertension – a sequelae of chronic liver disease. The spleen is enlarged due to increased blood flow as a result of a stiffened cirrhotic liver (which is resistant to flow and therefore the blood takes the path of least resistance – see discussion below).
Neonatal liver disease
Jaundice in the neonatal period is common. About two-thirds of term babies have transient jaundice 3–5 days after birth. Most do not have liver disease. Unconjugated jaundice is often due to immaturity of the hepatic enzyme glucuronosyltransferase, responsible for glucuronidation of bilirubin. Unconjugated jaundice in the neonatal period may be due to ‘breastmilk jaundice’. Other causes include haemolysis, sepsis and hypothyroidism. This is considered in detail in Chapter 11, Neonatal medicine. In contrast to this, conjugated hyperbilirubinaemia is often a reflection of hepatic dysfunction due to a number of potential causes, including biliary atresia or neonatal hepatitis syndrome.
Babies with significant liver problems usually have significant jaundice. Under these circumstances, the bilirubin level should be measured together with the fractional breakdown of the conjugated and unconjugated components. This will help to guide further investigation and management.
Disorders of bilirubin metabolism
Crigler–Najjar syndrome types I and II are rare autosomal recessive disorders which cause intense unconjugated hyperbilirubinaemia in the first days of life, which persists thereafter. In type I, there is no UDP-glucuronosyltransferase. Hyperbilirubinaemia is very severe despite phototherapy and may result in kernicterus. Diagnosis is made by DNA analysis. In type I, there is no response to treatment with phenobarbital, which causes cytochrome P450 enzyme induction. Acute treatment is with exchange transfusion and liver transplantation is a long-term option. In type II, some UDP-glucuronosyltransferase is present, so hyperbilirubinaemia is less severe. Phenobarbital therapy is effective, generally with a marked decrease in serum bilirubin. Therapies based on gene and cell transfer techniques are likely to be helpful in the future.
Question 21.3
Bilrubin metabolism
A six-week-old male baby is referred to the general paediatric clinic as a ‘hungry baby who cries all the time’. Upon review, the baby is alarmingly malnourished and jaundiced. The stool is white (Fig. 21.4) and urine dark orange. Which of the following is most likely to BEST describe the physiology underpinning the pale stool? Select ONE answer only. The stool is pale because it is lacking in:
Conjugated neonatal jaundice
In normal circumstances, only a small fraction of bilirubin is conjugated. In neonates, conjugated hyperbilirubinaemia is defined as a serum conjugated bilirubin of greater than 25% of the total (current BSPGHAN guideline) or >25 µmol/L (NICE guidelines). This most commonly occurs when there is cholestasis. With neonatal cholestasis, there is impairment of bile excretion caused by defects in intrahepatic production or transmembrane transport of bile, or mechanical obstruction to bile flow. The biochemical features of cholestasis reflect the retention of components of bile in the serum (bilirubin, bile acids, and/or cholesterol). The pattern and severity of each of these abnormalities varies with the underlying disorder.
Classically, patients present with jaundice, pale (acholic) stool and dark urine. Pale stools occur as no bilirubin reaches the gastrointestinal tract and dark urine results from excretion of water-soluble conjugated bilirubin in the urine. Both of these features suggest cholestasis.
Biliary atresia
Biliary atresia is the commonest cause of neonatal liver disease and indication for liver transplantation in children. It occurs in 1 : 17,000 live births. It presents with conjugated jaundice, acholic stools and hepatomegaly. There is failure of passage of bile from the liver into the gall bladder and onto the small intestine. Bile is essential for carrying waste from the liver and promoting absorption of fats and fat-soluble vitamins. Biliary atresia results in chronic liver failure if surgical correction is not performed. The Kasai portoenterostomy attempts to allow diversion of bile from the residual small bile ducts. The surgery involves attaching the porta hepatis to a loop of small intestine. Up to 60% will achieve biliary drainage (bilirubin <20 µmol/L) within 6 months. Most of these children will reach adolescence with a good quality of life (but with cirrhosis/evidence of portal hypertension) without undergoing liver transplantation.
Congenital infection
Congenital infections are described in detail in Chapter 10, Perinatal medicine. The clinical features of congenital CMV, toxoplasmosis, rubella and syphilis include conjugated jaundice and hepatomegaly. However, any infection acquired around the time of birth, e.g. herpes simplex, varicella zoster virus or any bacterial infection, can cause cholestasis as hepatic bile flow is very sensitive to circulating endotoxins.
Genetically inherited cholestatic liver disease
The liver plays a number of important roles in amino acid metabolism, protein synthesis, carbohydrate metabolism and lipid metabolism. It is also the site of manufacture of a number of blood coagulation proteins. It is therefore key to much of the metabolic activity in the body and over 100 heritable forms of liver disease have been described. The clinical features and genetics of some of the genetically inherited diseases affecting the liver are summarized in Table 21.2.
Table 21.2
Some of the commoner genetic diseases affecting the liver presenting with conjugated hyperbilirubinaemia
| Name | Frequency | Genetics | Associated features | Treatment |
| Alpha-1-antitrypsin deficiency | 1 : 1600 | AR (SERPINA1 gene) 3 alleles: M, S and Z. Most common deficiency is Z-amino acid substitution. | Presentation in neonatal period is with conjugated jaundice, acholic stools and intrauterine growth restriction. Variable hepatomegaly. Hepatitis, cirrhosis and liver failure. | Cirrhosis and risk of malignant transformation. Family screening. Alcohol and smoking advice. Liver transplantation for chronic liver disease. |
| Alagille syndrome | 1 : 100,000 | AD with incomplete penetrance, due to defects in the JAG1 gene on chromosome 20 coding for the ligand Notch1. | The expression of Notch1 found in many of the organs, e.g. liver, kidney and heart. Cardiac, renal and dysmorphic features common, i.e. characteristic facies (broad forehead, hypertelorism, deep set eyes, and small pointed chin). Butterfly vertebrae due to fusion of anterior arch of vertebral body are commonly found in the thoracic spine. | Supportive with vitamins. Medication to control intense pruritus. May need transplantation for chronic liver disease. |
| Tyrosinaemia type 1 |



