Chapter 456 Hemoglobinopathies
456.1 Sickle Cell Disease
Clinical Manifestations and Treatment of Sickle Cell Anemia
Fever and Bacteremia
Fever in a child with sickle cell anemia is a medical emergency, requiring prompt medical evaluation and delivery of antibiotics due to the increased risk of bacterial infection and concomitant high fatality rate with infection. Several clinical management strategies have been developed for children with fever, ranging from admitting all patients with a fever for IV antimicrobial therapy to administering a 3rd-generation cephalosporin in an outpatient setting to patients without any of the previously established risk factors for occult bacteremia (Table 456-1). Given the observation that the average time for a positive blood culture with a bacterial pathogen is <20 hr in children with sickle cell anemia, admission for 24 hr is probably the most prudent strategy for children and families without a telephone or transportation, or with a history of inadequate follow-up. Outpatient management should be considered only for those with the lowest risk for bacteremia, and treatment choice should be considered carefully.
Table 456-1 CLINICAL FACTORS ASSOCIATED WITH INCREASED RISK OF BACTEREMIA REQUIRING ADMISSION IN FEBRILE CHILDREN WITH SICKLE CELL DISEASE
Seriously ill appearance
Hypotension: systolic BP <70 mm Hg at 1 year of age or <70 mm Hg + 2 × the age in yr for older children
Poor perfusion: capillary-refill time >4 sec
Temperature >40.0°C
A corrected white-cell count >30,000/mm3 or <500/mm3
Platelet count <100,000/mm3
History of pneumococcal sepsis
Severe pain
Dehydration: poor skin turgor, dry mucous membranes, history of poor fluid intake, or decreased output of urine
Infiltration of a segment or a larger portion of the lung
Hemoglobin level <5.0 g/dL
BP, blood pressure.
From Williams JA, Flynn PM, Harris S et al: A randomized study of outpatient treatment with ceftriaxone for selected febrile children with sickle cell disease, N Engl J Med 329:472–476, 1993.
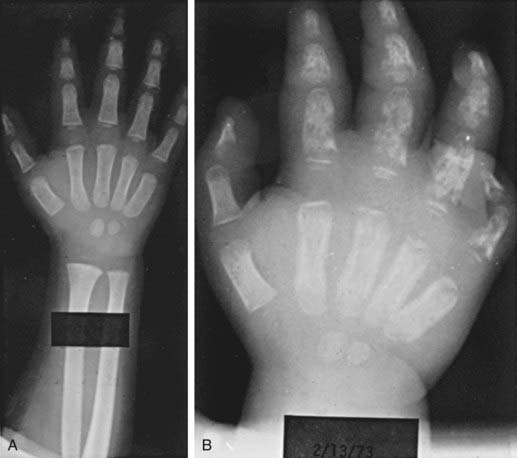
Dactylitis
Dactylitis, often referred to as hand-foot syndrome, is often the first manifestation of pain in children with sickle cell anemia, occurring in 50% of children by their 2nd year (Fig. 456-1). Dactylitis often manifests with symmetric or unilateral swelling of the hands and/or feet. Unilateral dactylitis can be confused with osteomyelitis, and careful evaluation to distinguish between the two is important, because treatment differs significantly. Dactylitis requires palliation with pain medications, such as acetaminophen with codeine, whereas osteomyelitis requires at least 4-6 wk of IV antibiotics.
Pain
Some patients require hospitalization for administration of IV morphine or derivatives of morphine. The incremental increase and decrease in the use of the medication to relieve pain roughly parallels the 8 phases associated with a chronology of pain and comfort (Table 456-2). The average hospital length of stay for children admitted in pain is 4.4 days. The American Pain Society has published clinical guidelines for treating acute and chronic pain in patients with sickle cell disease of any type. These recommendations are comprehensive and represent a starting point for treating pain (www.ampainsoc.org/pub/sc.htm).
Table 456-2 SUMMARY OF THE CHRONOLOGY OF PAIN IN CHILDREN WITH SICKLE CELL DISEASE
| PHASE | PAIN CHARACTERISTICS | SUGGESTED COMFORT MEASURES USED |
|---|---|---|
| 1 (Baseline) | No vaso-occlusive pain; pain of complications may be present, such as that connected with avascular necrosis of the hip | No comfort measures used |
| 2 (Pre-pain) | No vaso-occlusive pain; pain of complications may be present; prodromal signs of impending vaso-occlusive episode may appear, e.g., “yellow eyes” and/or fatigue | No comfort measures used; caregivers may encourage child to increase fluids to prevent pain event from occurring |
| 3 (Pain start point) | First signs of vaso-occlusive pain appear, usually in mild form | Mild oral analgesic often given; fluids increased; child usually maintains normal activities |
| 4 (Pain acceleration) | Intensive of pain increases from mild to moderate Some children skip this level or move quickly from phase 3 to phase 5 | Stronger oral analgesic are given; rubbing, heat, or other activities are often used; child usually stays in school until the pain becomes more severe, then stays home and limits activities; is usually in bed; family searches for ways to control the pain |
| 5 (Peak pain experience) | Pain accelerates to high moderate or severe levels and plateaus; pain can remain elevated for extended period Child’s appearance, behavior, and mood are significantly different from normal | Oral analgesics are given around the clock at home; combination of comfort measures is used; family might avoid going to the hospital; if pain is very distressing to the child, parent takes the child to the emergency department After child enters the hospital, families often turn over comforting activities to health care providers and wait to see if the analgesics work Family caregivers are often exhausted from caring for the child for several days with little or no rest |
| 6 (Pain decrease start point) | Pain finally begins to decrease in intensity from the peak pain level | Family caregivers again become active in comforting the child but not as intensely as during phases 4 and 5 |
| 7 (Steady pain decline) | Pain decreases more rapidly, become more tolerable for the child Child and family are more relaxed | Health care providers begin to wean the child from the IV analgesic; oral opioids given; discharge planning is started Children may be discharged before they are pain free |
| 8 (Pain resolution) | Pain intensity is at a tolerable level, and discharge is imminent Child looks and acts like “normal” self Mood improves | May receive oral analgesics |
Adapted from Beyer JE, Simmons LE, Woods GM, et al: A chronology of pain and comfort in children with sickle cell disease, Arch Pediatr Adolesc Med 153:913–920, 1999.
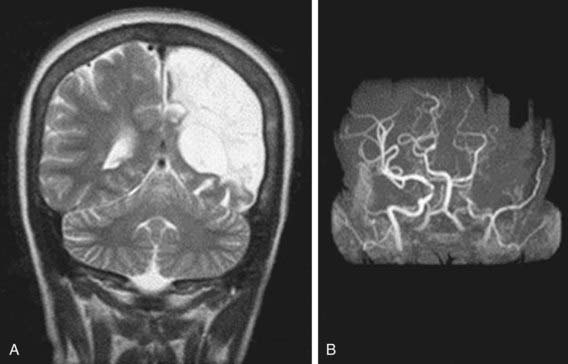
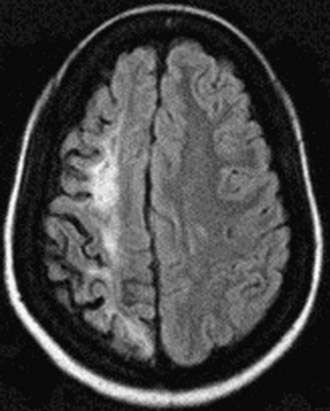
Neurologic Complications
Neurologic complications associated with sickle cell anemia are varied and complex. Approximately 11% and 20% of children with sickle cell anemia will have overt and silent strokes, respectively, before their 18th birthday (Figs. 456-2 and 456-3). An overt stroke is defined as a focal neurologic deficit lasting >24 hr. However, this definition is outdated because many patients with sickle cell anemia will be treated with blood therapy that can hasten their recovery to baseline. A more functional definition is the presence of a focal neurologic deficit that lasts for >24 hr and/or increased signal intensity with a T2-weighted MRI of the brain indicating a cerebral infarct, corresponding to the focal neurologic deficit. The definition of silent cerebral infarct is the absence of a focal neurologic deficit lasting >24 hr in the presence of a lesion on T2-weighted MRI indicating a cerebral infarct. Evidence of a stroke can be found as early as 1 yr of age. Other neurologic complications include headaches that may or may not be related to sickle cell anemia, seizures, cerebral venous thrombosis, and reversible posterior leukoencephalopathy syndrome (RPLS). Children with other types of sickle cell disease such as Hb SC or Hb Sβ-thalassemia plus might have overt or silent cerebral infarcts as well.
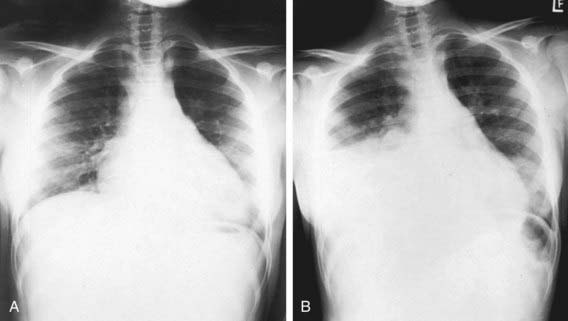
Lung Disease
Lung disease in children with sickle cell anemia is the second most common reason for admission to the hospital and a common cause of death. ACS refers to a constellation of findings that include a new radiodensity on chest radiograph, fever, respiratory distress, and pain that occurs often in the chest, but it can also include the back and/or abdomen only (Fig. 456-4). Even in the absence of respiratory symptoms, all patients with fever should receive a chest radiograph to identify ACS because clinical examination alone is insufficient to identify patients with a new radiographic density, and early detection of acute syndrome will alter clinical management. The radiographic findings in ACS are variable but can include involvement of a single lobe (predominantly the left lower lobe) or multiple lobes (most often both lower lobes) and pleural effusions (either unilateral or bilateral).
Given the clinical overlap between ACS and common pulmonary complications such as bronchiolitis, asthma, and pneumonia, a wide range of therapeutic strategies have been used (Table 456-3). Oxygen administration and blood transfusion therapy, either simple or exchange (manual or automated), are the most common interventions used to treat ACS. Supplemental oxygen should be administered when the room air oxygen saturation is >90%. The decision about when to give blood and whether the transfusion should be a simple or exchange transfusion is less clearly defined. Commonly, blood transfusions are given when at least one of the following clinical features is present: decreasing oxygen saturation, increase work of breathing, rapid change in respiratory effort either with or without a worsening chest radiograph, or previous history of severe ACS requiring admission to the intensive care unit.
Table 456-3 OVERALL STRATEGIES FOR THE MANAGEMENT OF ACUTE CHEST SYNDROME
PREVENTION
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


