Hematology
Nirali Shah
Eric F. Grabowski
Anemia
Definition
A reduction of RBC volume or hemoglobin conc < range of nml in healthy pts
Hematologic parameters vary w/ age (interpret using age-specific indices)
Microcytic anemia is the most common category of anemia in children
Clinical Manifestations
Can be asymptomatic or w/ fatigue, irritability, dyspnea
History: Diet Hx: Nutritional iron def is primary cause of microcytic anemia in children
Pica, Pagophagia (Craving and eating ice; common with iron deficiency)
FHx: Eval hemoglobinopathy in African Americans, Mediterranean region/southeast Asia
Signs or symptoms of blood loss: Hematochezia, melena, heavy menses
Exam: Pallor, tachycardia/flow murmur, assess for splenomegaly
Diagnostic Studies
CBC, MCV, retic, hemolysis labs (Coombs, bilirubin, haptoglobin, LDH), smear
Hemoglobin and mean corpuscular volume by age. (J Pediatr 1979;94:26)
Ex: 6 mo MCV nml range 71–94 fL, 18 yo MCV nml range 78–98 fL
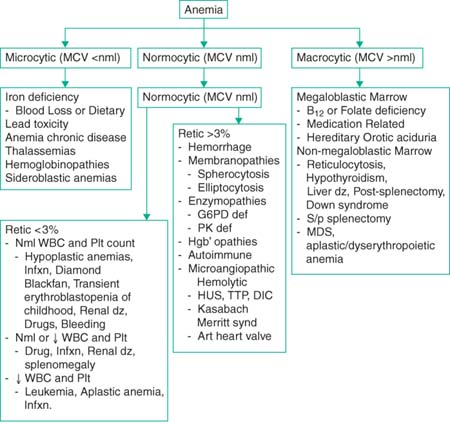 |
Microcytic Anemia
(Pediatr Rev 2007;28:5; N Engl J Med 1999;341:1986)
Mentzer index (MCV/RBC): >13 consistent w/ iron def; <13 with beta-thal trait
Iron studies:
Iron deficiency: ↓ Iron ↑ TIBC ↓ Ferritin
Chronic disease: ↓ Iron ↓ TIBC ↑ Ferritin
Thalassemia/lead: ↑-Nml Iron ↓ TIBC ↑ Ferritin
Iron deficiency anemia: Most common form of microcytic anemia
Rx: 6 mg/kg elemental Fe ×6 wk in 2–3 divided doses, IV iron in severe def, blood transfusion. Vitamin C increases absorption of iron.
Tea, phytates (e.g., in corn) diminish iron absorption.
Typically improvement in reticulocyte count, MCV, and hemoglobin in 1–4 wk
Anemia of chronic disease: Can be microcytic or normocytic. (N Engl J Med 2005;352:1011); hepcidin (↑ w/ inflam) blocks Fe release
Treat underlying disorder (i.e., epo for renal disease)
Normocytic Anemia
(Pediatr Rev 1988;10:77)
Low retic: Diamond-Blackfan, trans erythroblastopenia of childhood, aplastic crisis
High retic: Hemolysis (including autoimmune), G6PD, hereditary spherocytosis
Macrocytic Anemia
(Ped Rev 1988;10:77; N Engl J Med 1999;341:1986)
Vit B12 deficiency: Rare in kids, 2/2 pernicious anemia and ileal disease (Crohns)
Assoc w/ pancytopenia w/ macrocytosis, hyperseg PMNs
Exam: Glossitis and decreased vibration and position senses
Neuro manifestations → can be irreversible if untreated
Rx: Oral or parenteral Vitamin B12 supplementation
Folate deficiency: Rare 2/2 ↑ in folate supplementation for pregnancy
Found in malabsorption syndromes, EtOH use, chronic hemolysis, drugs (MTX)
R/o Vit B12 def (folate supps fix RBC parameters; B12 def can be missed)
Hemoglobinopathies
Sickle Cell Disease (SCD)
(Pediatr Rev 2007;28:259)
Definition: Chronic hemolytic anemia; includes Hgb variants SS, SC, S-beta thal, SO Arab, SD, and other rare S-Hb genotypes
SS dz: Both β-globin genes w/ mutation (valine for glutamate at AA 6 on β-chain)
SC dz: 1 β-globin chain w/ mutation (lysine for glutamate at AA 6)
CC disease: Relatively mild, mild microcytic anemia
Sickle-β-thal: 1 β-globin gene w/ S mut, other nonfxnl. Similar course as SS dz
Pathophysiology:
Nucleotide sub (GTG for GAG; codon 6) of β-globin gene (chromo 11); valine for glut acid; HbS polymerizes on deoxygenation, distorts RBC into crescent
Sickled cells less deformable and ↑ adherence to vasc endothelium; lead to vascular occlusion, organ ischemia, and chronic end organ damage.
Epidemiology:
African American: 1 in 375, 9% carrier prev in U.S.; Hispanics: 1 in 1200
Clinical manifestations: Appear in first yr as fetal Hb concentrations decline
Fetal Hb protective because it inhibits deoxy-Hb S polymerization in the RBCs
Those w/ persistence of fetal Hb (HbF >30%) have mild or no symptoms
See later in chapter
Diagnostic studies: Newborn screening (NS) performed in 44 states and DC
If NS not offered, high risk infants tested by electrophoresis before 3 mo
Sickledex (rapid test based on solubility) inappropriate in newborns
Prophylaxis: PCN, pneumococcal vaccine, hydroxyurea
Transcranial Doppler (TCD): If flow velocity >200 cm/sec, ↑ risk for stroke. Chronic transfxn therapy ↓ 1° stroke 10×’s in Hb-SS and Hb-S-β.
Screening all kids w/ Hb-SS and Hb-S-β btw 2–16 yo. D/c of Rx after 36 mo of Rx resulted in reversion to abn TCD velocity or stroke in 45% of pts.
Recs for indefinite transfusion Rx for pts who have abn TCD findings
Complications and Management
| Complication | Features | Evaluation & Treatment |
|---|---|---|
| Infection | Fever >101.5 | CBC/diff, retic, U/A, pan cx, CXR. CFTX, +/- Vanco Outpt if: well appearing, >6 mo, WBC <25, nl CXR, no UTI, Tm <103, and reliable f/u |
| – S. pneumo sepsis | Children <5 | Ceftriaxone (CFTX) |
| – Osteomyelitis | Salmonella, Staph. Aureus | Hard to differentiate from VOC (see below), ESR, blood and bone aspirate |
| – Parvo B19 | Aplastic crisis | Transfusion. Reticulocytopenia 7–10 d. Immunity is lifelong. |
| Splenic sequestration | Hb-SS <3 yr; Hb-SC, S-β-thal any age. Sudden splenomegaly and >2 g/dL ↓ Hgb from baseline w/ ↑ retics. | CBC/diff, monitor spleen tip. Transfusion as needed |
| Pulmonary Features (N Engl J Med 2008;359:2254) | ||
| Acute chest syndrome | 50% pts (#2 cause of admission). Defined by CXR w/ new pulmonary infiltrate w/ fever &/or resp sx’s. Hypoxia not necessary for dx. | CFTX/Azithro, xfuse w/ pRBC if O2 <95%, or Hct <18%, do not exceed Hct >35% 2/2 viscosity. IV hydrate NOT more than 1 × maintenance, IS, albuterol nebs |
| Pulmonary HTN | 30% of adults who have Hb-SS | Optimal management still under investigation |
| Vaso-Occlusive pain crisis (VOC) | 70% of all patients; 5% (Hb-SS) account for 30% of admissions (#1 cause of hospitalizations) | NS bolus 20 cc/kg and hydrate at 1.5 × M (unless ACS); morphine PCA, Toradol, Incent spir (IS), bowel reg |
| Anemia | Tranfuse w/ Sickle negative, CMV safe, leukoreduced, extended phenotype cross matched | Transfuse for Hgb <5; falling Hct with low retic; ACS w/O2 requirement; stroke; splenic sequestration |
| Neurologic Features | ||
| Overt stroke | 11% of Hb-SS by age 20 yr | Xfuse if fixed or evolving deficit, focal szr, exchange xfusion, or RBC pheresis if >20 kg to ↓ HBS <30% |
| Silent infarction MRI | 22% of Hb-SS children | |
| Osteonecrosis—Femoral/humoral head | 50% of Hb-SS (increased with alpha-thalassemia); 60% of Hb-SC | |
| Retinopathy | 50% of Hb-SC adults | Routine ophthalmology visits |
| Renal insufficiency | 5%–20% of Hb-SS adults | |
| Cholelithiasis | 42% by adolescence | |
| Leg ulcers | 10%–25% of Hb-SS adults | |
| Priapism | 10%–40% of Hb-SS males | Urology consult |
Thalassemias
(Pediatr Rev 2002;23:75; Pediatr Rev 2007;28:5)
Nml Hgb (Hgb A) composed of 2 β-chains and 2 α-chains.
Fetal Hgb (Hgb F) composed of 2 γ-chains and 2 α-chains.
Consider dx w/ hypochromic, microcytic anemia, and in pts of appropriate ethnic background
Beta-Thalassemia Trait
AR, β-chains controlled by two genes on chromosome 11; no Rx needed
Diminished production of nml β-globin chains → hypochromic, microcytic anemia
Characterized by mild anemia (hemoglobin 9.5–11) and MCV <80
Basophilic stippling often seen on blood smear
Beta-Thalassemia Major
(Cooley Anemia)
Little to no production of beta-chains, and subsequently hemoglobin A (alpha2, beta2)
Usually evident beyond 6 mo of age when fetal hemoglobin production falls
Severe hypochromic, microcytic anemia requires chronic transfusion
Enlargement of liver and spleen because of extramedullary hematopoiesis
Long-term complications those of iron overload; BMT from matched donor curative
Alpha-Thalassemia
Normal: 4 functional genes for alpha-chain (on chromosome 16)
Silent carrier: 3 functional genes
α-thalassemia trait: 2 functional genes, typically asymptomatic w/ ↓ Hgb
Hgb H: 1 functional gene. Hgb H composed of tetrad of β-chains 2/2 paucity of available α-chains. Characterized by chronic hemolytic anemia w/ splenomegaly.
Bart Hgb: 0 functional genes. Bart Hgb: tetrad of four γ-chains, leading to catastrophic anemia, hydrops fetalis, and fetal or neonatal loss.
Platelet Disorders
Thrombocytopenia
(Pediatr Rev 2005;26:401)
Definition: Plt count <150,000/μL
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


