FIGURE 56-1 Mean hemoglobin concentrations (black line) and 5th and 95th percentiles (blue lines) for healthy pregnant women taking iron supplements. (Data from the Centers for Disease Control and Prevention, 1989.)
The modest fall in hemoglobin levels during pregnancy is caused by a relatively greater expansion of plasma volume compared with the increase in red cell volume (Chap. 4, p. 55). The disproportion between the rates at which plasma and erythrocytes are added to the maternal circulation is greatest during the second trimester. Late in pregnancy, plasma expansion essentially ceases, while hemoglobin mass continues to increase.
 General Considerations
General Considerations
The causes of anemia in pregnancy and their frequency are dependent on multiple factors such as geography, ethnicity, nutritional status, preexisting iron status, and prenatal iron supplementation. Other factors are socioeconomic, and anemia is more prevalent among indigent women (American College of Obstetricians and Gynecologists, 2013a). Approximately 25 percent of almost 48,000 Israeli pregnant women had a hemoglobin level < 10 g/dL (Kessous, 2013). Ren and colleagues (2007) found that 22 percent of 88,149 Chinese women were anemic in the first trimester. Of 1000 Indian women, half were anemic at some point, and 40 percent were throughout pregnancy (Kumar, 2013). The importance of prenatal iron therapy is illustrated by the study of Taylor and associates (1982), who reported that hemoglobin levels at term averaged 12.7 g/dL among women who took supplemental iron compared with 11.2 g/dL for those who did not. Bodnar and coworkers (2001) studied a cohort of 59,248 pregnancies and found a postpartum anemia prevalence of 27 percent that correlated both with prenatal anemia and hemorrhage at delivery.
The etiologies of the more common anemias encountered in pregnancy are listed in Table 56-1. The specific cause of anemia is important when evaluating effects on pregnancy outcome. For example, maternal and perinatal outcomes are seldom affected by moderate iron deficiency anemia, yet they are altered markedly in women with sickle-cell anemia.
TABLE 56-1. Causes of Anemia During Pregnancy
Acquired
Iron deficiency anemia
Anemia caused by acute blood loss
Anemia of inflammation or malignancy
Megaloblastic anemia
Acquired hemolytic anemia
Aplastic or hypoplastic anemia
Hereditary
Thalassemias
Sickle-cell hemoglobinopathies
Other hemoglobinopathies
Hereditary hemolytic anemias
 Effects on Pregnancy Outcomes
Effects on Pregnancy Outcomes
Most studies of anemia during pregnancy describe large populations. As indicated, these likely deal with nutritional anemias, specifically those due to iron deficiency. Klebanoff and associates (1991) studied nearly 27,000 women and found a slightly increased risk of preterm birth with midtrimester anemia. Ren and colleagues (2007) found that a low first-trimester hemoglobin concentration increased the risk of low birthweight, preterm birth, and small-for-gestational age infants. In a study from Tanzania, Kidanto and coworkers (2009) reported that the incidence of preterm delivery and low birthweight was increased as the severity of anemia increased. They did not, however, take into account the cause(s) of anemia, which was diagnosed in almost 80 percent of their obstetrical population. Kumar and colleagues (2013) studied 1000 Indian women and also found that second- and third-trimester anemia was associated with preterm birth and low birthweight. Chang and associates (2013) followed 850 children born to women classified as iron deficient in the third trimester. Children without iron supplementation had lower mental development at 12, 18, and 24 months of age, suggesting that prenatal iron deficiency is associated with mental development. Tran and associates (2014) reported similar findings from a Vietnamese study.
A seemingly paradoxical finding is that healthy pregnant women with a higher hemoglobin concentration are also at increased risk for adverse perinatal outcomes (von Tempelhoff, 2008). This may result from lower than average plasma volume expansion of pregnancy concurrent with normal red cell mass increase. Murphy and coworkers (1986) described more than 54,000 singleton pregnancies in the Cardiff Birth Survey and reported excessive perinatal morbidity with high maternal hemoglobin concentrations. Scanlon and associates (2000) studied the relationship between maternal hemoglobin levels and preterm or growth-restricted infants in 173,031 pregnancies. Women whose hemoglobin concentration was three standard deviations above the mean at 12 or 18 weeks’ gestation had 1.3- to 1.8-fold increases in the incidence of fetal-growth restriction. These findings have led some to the illogical conclusion that withholding iron supplementation to cause iron deficiency anemia will improve pregnancy outcomes (Ziaei, 2007).
 Iron Deficiency Anemia
Iron Deficiency Anemia
The two most common causes of anemia during pregnancy and the puerperium are iron deficiency and acute blood loss. The CDC (1989) estimated that as many as 8 million American women of childbearing age were iron deficient. In a study of more than 1300 women, 21 percent had third-trimester anemia with 16 percent due to iron deficiency anemia (Vandevijvere, 2013). In a typical singleton gestation, the maternal need for iron averages close to 1000 mg. Of this, 300 mg is for the fetus and placenta; 500 mg for maternal hemoglobin mass expansion; and 200 mg that is shed normally through the gut, urine, and skin. The total amount of 1000 mg considerably exceeds the iron stores of most women and results in iron deficiency anemia unless iron supplementation is given.
Iron deficiency is often manifested by an appreciable drop in hemoglobin concentration. In the third trimester, additional iron is needed to augment maternal hemoglobin and for transport to the fetus. Because the amount of iron diverted to the fetus is similar in a normal and in an iron-deficient mother, the newborn infant of a severely anemic mother does not suffer from iron deficiency anemia. As discussed in Chapter 33 (p. 643), neonatal iron stores are related to maternal iron status and to timing of cord clamping.
Diagnosis
Classic morphological evidence of iron deficiency anemia—erythrocyte hypochromia and microcytosis—is less prominent in the pregnant woman compared with that in the nonpregnant woman. Moderate iron deficiency anemia during pregnancy usually is not accompanied by obvious morphological changes in erythrocytes. Serum ferritin levels, however, are lower than normal, and there is no stainable bone marrow iron. Iron deficiency anemia during pregnancy is the consequence primarily of expansion of plasma volume without normal expansion of maternal hemoglobin mass.
The initial evaluation of a pregnant woman with moderate anemia should include measurements of hemoglobin, hematocrit, and red cell indices; careful examination of a peripheral blood smear; a sickle-cell preparation if the woman is of African origin; and measurement of serum iron or ferritin levels, or both. Expected values in pregnancy are found in the Appendix (p. 1287). Serum ferritin levels normally decline during pregnancy (Goldenberg, 1996). Levels less than 10 to 15 mg/L confirm iron deficiency anemia (American College of Obstetricians and Gynecologists, 2013a). Pragmatically, the diagnosis of iron deficiency in moderately anemic pregnant women usually is presumptive and based largely on exclusion.
When pregnant women with moderate iron deficiency anemia are given adequate iron therapy, a hematological response is detected by an elevated reticulocyte count. The rate of increase of hemoglobin concentration or hematocrit is typically slower than in nonpregnant women due to the increasing and larger blood volumes during pregnancy.
Treatment
Regardless of anemia status, daily oral supplementation with 30 to 60 mg of elemental iron and 400 μg of folic acid is recommended in pregnancy (Peña-Rosas, 2012; World Health Organization, 2012). Anemia resolution and restitution of iron stores can be accomplished with simple iron compounds—ferrous sulfate, fumarate, or gluconate—that provide about 200 mg daily of elemental iron. If a woman cannot or will not take oral iron preparations, then parenteral therapy is given. Although both are administered intravenously, ferrous sucrose has been shown to be safer than iron-dextran (American College of Obstetricians and Gynecologists, 2013a). There are equivalent increases in hemoglobin levels in women treated with either oral or parenteral iron therapy (Bayouneu, 2002; Sharma, 2004).
 Anemia from Acute Blood Loss
Anemia from Acute Blood Loss
In early pregnancy, anemia caused by acute blood loss is common with abortion, ectopic pregnancy, and hydatidiform mole. Anemia is much more common postpartum from obstetrical hemorrhage. Massive hemorrhage demands immediate treatment as described in Chapter 41 (p. 814). If a moderately anemic woman—defined by a hemoglobin value of approximately 7 g/dL—is hemodynamically stable, is able to ambulate without adverse symptoms, and is not septic, then blood transfusions are not indicated. Instead, iron therapy is given for at least 3 months (Krafft, 2005). In a randomized trial, Van Wyck and colleagues (2007) reported that intravenous ferric carboxymaltose given weekly was as effective as thrice-daily oral ferrous sulfate tablets for hemoglobin regeneration for postpartum anemia.
 Anemia Associated with Chronic Disease
Anemia Associated with Chronic Disease
Weakness, weight loss, and pallor have been recognized since antiquity as characteristics of chronic disease. Various disorders, such as chronic renal failure, cancer and chemotherapy, human immunodeficiency virus (HIV) infection, and chronic inflammation, result in moderate and sometimes severe anemia, usually with slightly hypochromic and microcytic erythrocytes. It is the second most common form of anemia worldwide (Weiss, 2005).
In nonpregnant patients with chronic inflammatory diseases, the hemoglobin concentration is rarely < 7 g/dL; bone marrow cellular morphology is not altered; and serum iron concentrations are decreased. However, ferritin levels usually are elevated. The low levels of iron are mediated by hepcidin, a polypeptide produced in the liver that participates in iron balance and transport (Weiss, 2005). These anemias share similar features that include alterations in reticuloendothelial function, iron metabolism, and decreased erythropoiesis (Cullis, 2013).
Pregnancy
Women with chronic disorders may develop anemia for the first time during pregnancy. In those with preexisting anemia, it may be intensified as plasma volume expands disproportionately to red cell mass expansion. Causes include chronic renal insufficiency, inflammatory bowel disease, and connective-tissue disorders. Others are granulomatous infections, malignant neoplasms, rheumatoid arthritis, and chronic suppurative conditions.
Of these, chronic renal insufficiency is the most common disorder that we have encountered as a cause of anemia during pregnancy. Some cases are accompanied by erythropoietin deficiency. As discussed in Chapter 53 (p. 1060), during pregnancy in women with mild chronic renal insufficiency, the degree of red cell mass expansion is inversely related to renal impairment. At the same time, plasma volume expansion usually is normal, and thus anemia is intensified (Cunningham, 1990). Anemia often accompanies acute pyelonephritis but is due to acute endotoxin-mediated erythrocyte destruction. With normal erythropoietin production, red cell mass is restored as pregnancy progresses (Cavenee, 1994; Dotters-Katz, 2013).
Treatment
Adequate iron stores must be ensured. Recombinant erythropoietin has been used successfully to treat chronic anemia (Weiss, 2005). In pregnancies complicated by chronic renal insufficiency, recombinant erythropoietin is usually considered when the hematocrit approximates 20 percent (Ramin, 2006). Cyganek and coworkers (2011) described good results in five pregnant renal transplant recipients treated with human recombinant erythropoietin. One worrisome side effect is hypertension, which is already prevalent in women with renal disease. In addition, Casadevall and colleagues (2002) reported pure red cell aplasia and antierythropoietin antibodies in 13 nonpregnant patients given recombinant human erythropoietin. Numerous additional cases have been reported. However, because of changes in manufacturing and new regulations, it is an infrequent toxicity today (McKoy, 2008).
 Megaloblastic Anemia
Megaloblastic Anemia
These anemias are characterized by blood and bone-marrow abnormalities from impaired DNA synthesis. Worldwide, the prevalence of megaloblastic anemia during pregnancy varies considerably. In the United States, it is rare.
Folic Acid Deficiency
In the United States, megaloblastic anemia beginning during pregnancy almost always results from folic acid deficiency. In the past, this condition was referred to as pernicious anemia of pregnancy. It usually is found in women who do not consume fresh green leafy vegetables, legumes, or animal protein. As folate deficiency and anemia worsen, anorexia often becomes intense and further aggravates the dietary deficiency. In some instances, excessive ethanol ingestion either causes or contributes to folate deficiency.
In nonpregnant women, the folic acid requirement is 50 to 100 μg/day. During pregnancy, requirements are increased, and 400 μg/day is recommended (Chap. 9, p. 181). The earliest biochemical evidence is low plasma folic acid concentrations (Appendix, p. 1287). Early morphological changes usually include neutrophils that are hypersegmented and newly formed erythrocytes that are macrocytic. With preexisting iron deficiency, macrocytic erythrocytes cannot be detected by measurement of the mean corpuscular volume. Careful examination of a peripheral blood smear, however, usually demonstrates some macrocytes. As the anemia becomes more intense, peripheral nucleated erythrocytes appear, and bone marrow examination discloses megaloblastic erythropoiesis. Anemia may then become severe, and thrombocytopenia, leukopenia, or both may develop. The fetus and placenta extract folate from maternal circulation so effectively that the fetus is not anemic despite severe maternal anemia. There have been instances in which newborn hemoglobin levels were 18 g/dL or more, whereas maternal values were as low as 3.6 g/dL (Pritchard, 1970). A Cochrane review by Lassi and associates (2013) evaluated the effectiveness of oral prenatal folic acid supplementation alone or with other micronutrients versus no folic acid. There was no conclusive evidence of supplement benefit for pregnancy outcomes that included preterm birth and perinatal mortality. There was, however, increased mean birthweight and a significant reduction in the incidence of megaloblastic anemia.
Treatment. Folic acid is given along with iron, and a nutritious diet is encouraged. As little as 1 mg of folic acid administered orally once daily produces a striking hematological response. By 4 to 7 days after beginning folic acid treatment, the reticulocyte count is increased, and leukopenia and thrombocytopenia are corrected.
Prevention. A diet sufficient in folic acid prevents megaloblastic anemia. The role of folate deficiency in the genesis of neural-tube defects has been well studied (Chap. 14, p. 283). Since the early 1990s, nutritional experts and the American College of Obstetricians and Gynecologists (2013c) have recommended that all women of childbearing age consume at least 400 μg of folic acid daily. More folic acid is given in circumstances in which requirements are increased. These include multifetal pregnancy, hemolytic anemia, Crohn disease, alcoholism, and inflammatory skin disorders. There is evidence that women who previously have had infants with neural-tube defects have a lower recurrence rate if a daily 4-mg folic acid supplement is given preconceptionally and throughout early pregnancy.
Vitamin B12 Deficiency
During pregnancy, vitamin B12 levels are lower than nonpregnant values because of decreased levels of binding proteins that include haptocorrin—transcobalamins I and III—and transcobalamin II (Morkbak, 2007). During pregnancy, megaloblastic anemia is rare from deficiency of vitamin B12, that is, cyanocobalamin. The typical example is Addisonian pernicious anemia, which results from absent intrinsic factor that is requisite for dietary vitamin B12 absorption. This autoimmune disorder usually has its onset after age 40 years (Stabler, 2013).
In our limited experience, vitamin B12 deficiency in pregnancy is more likely encountered following gastric resection. Those who have undergone total gastrectomy require 1000 μg of vitamin B12 given intramuscularly each month. Those with a partial gastrectomy usually do not need supplementation, but adequate serum vitamin B12 levels should be ensured during pregnancy (Appendix, p. 1290). Other causes of megaloblastic anemia from vitamin B12 deficiency include Crohn disease, ileal resection, and bacterial overgrowth in the small bowel (Stabler, 2013).
 Hemolytic Anemia
Hemolytic Anemia
There are several conditions in which accelerated erythrocyte destruction is stimulated by a congenital red cell abnormality or by antibodies directed against red cell membrane proteins. The cause may also go unproven. In some cases, hemolysis may be the primary disorder—some examples include sickle-cell disease and hereditary spherocytosis. In other cases, hemolysis develops secondary to an underlying disorder—examples include lupus erythematosus and the preeclampsia syndrome.
Autoimmune Hemolysis
The cause of aberrant antibody production in this uncommon condition is unknown. Typically, both the direct and indirect antiglobulin (Coombs) tests are positive. Anemias caused by these factors may be due to warm-active autoantibodies (80 to 90 percent), cold-active antibodies, or a combination. These syndromes also may be classified as primary (idiopathic) or secondary due to underlying diseases or other factors. Examples of the latter include lymphomas and leukemias, connective-tissue diseases, infections, chronic inflammatory diseases, and drug-induced antibodies (Provan, 2000). Cold-agglutinin disease may be induced by infectious etiologies such as Mycoplasma pneumoniae or Epstein-Barr viral mononucleosis (Dhingra, 2007). Hemolysis and positive antiglobulin test results may be the consequence of either IgM or IgG antierythrocyte antibodies. Spherocytosis and reticulocytosis are characteristic of the peripheral blood smear. When there is concomitant thrombocytopenia, it is termed Evans syndrome (Wright, 2013).
During pregnancy, there may be marked acceleration of hemolysis. This usually responds to glucocorticoids, and treatment is given with prednisone, 1 mg/kg given orally each day, or its equivalent. Coincidental thrombocytopenia usually is corrected by therapy. Transfusion of red cells is complicated by antierythrocyte antibodies, but warming the donor cells to body temperature may decrease their destruction by cold agglutinins.
Drug-Induced Hemolysis
These hemolytic anemias must be differentiated from other causes of autoimmune hemolysis. In most cases, hemolysis is mild, it resolves with drug withdrawal, and recurrence is prevented by avoidance of the drug. One mechanism is by hemolysis induced through drug-mediated immunological injury to red cells. The drug may act as a high-affinity hapten when bound to a red cell protein to which antidrug antibodies attach—for example, IgM antipenicillin or anticephalosporin antibodies. Some other drugs act as low-affinity haptens and adhere to cell membrane proteins—examples include probenecid, quinidine, rifampin, and thiopental. A more common mechanism for drug-induced hemolysis is related to a congenital erythrocyte enzymatic defect. An example is glucose-6-phosphate dehydrogenase (G6PD) deficiency, which is common in African American women (p. 1106).
Drug-induced hemolysis is usually chronic and mild to moderate, but occasionally there is severe acute hemolysis. For example, Garratty and coworkers (1999) described seven women with severe Coombs-positive hemolysis stimulated by cefotetan given as prophylaxis for obstetrical procedures. Alpha-methyldopa can cause similar hemolysis (Grigoriadis, 2013). Moreover, maternal hemolysis has been reported after intravenous immune globulin (IVIG) given for fetal and neonatal alloimmune thrombocytopenia (Rink, 2013). As treatment, response to glucocorticoids may be suboptimal, but withdrawal of the offending drug frequently halts the hemolysis.
Pregnancy-Induced Hemolysis
In some cases, unexplained severe hemolytic anemia develops during early pregnancy and resolves within months postpartum. There is no evidence of an immune mechanism or intraerythrocytic or extraerythrocytic defects (Starksen, 1983). Because the fetus-infant also may demonstrate transient hemolysis, an immunological cause is suspected. Maternal corticosteroid treatment is often—but not always—effective (Kumar, 2001). We have cared for a woman who during each pregnancy developed intense severe hemolysis with anemia that was controlled by prednisone. Her fetuses were not affected, and in all instances, hemolysis abated spontaneously after delivery.
Paroxysmal Nocturnal Hemoglobinuria
Although commonly regarded as a hemolytic anemia, this hemopoietic stem cell disorder is characterized by formation of defective platelets, granulocytes, and erythrocytes. Paroxysmal nocturnal hemoglobinuria is acquired and arises from one abnormal clone of cells, much like a neoplasm (Nguyen, 2006). One mutated X-linked gene responsible for this condition is termed PIG-A because it codes for phosphatidylinositol glycan protein A. Resultant abnormal anchor proteins of the erythrocyte and granulocyte membrane make these cells unusually susceptible to lysis by complement (Provan, 2000). The most serious complication is thrombosis, which is heightened in the hypercoagulable state of pregnancy.
Chronic hemolysis has an insidious onset, and its severity ranges from mild to lethal. Hemoglobinuria develops at irregular intervals and is not necessarily nocturnal. Hemolysis may be initiated by transfusions, infections, or surgery. Almost 40 percent of patients suffer venous thromboses and may also experience renal failure, hypertension, and Budd-Chiari syndrome. Because of the thrombotic risk, prophylactic anticoagulation is recommended (Parker, 2005). Median survival after diagnosis is 10 years, and bone marrow transplantation is the definitive treatment. Successful treatment of nonpregnant patients has been reported with eculizumab, an antibody that inhibits complement activation (Hillmen, 2006; Parker, 2009). Kelly and colleagues (2010) described seven pregnant women exposed to eculizumab with successful outcomes.
During pregnancy, paroxysmal nocturnal hemoglobinuria can be serious and unpredictable. Complications have been reported in up to three fourths of affected women, and the maternal mortality rate is 10 to 20 percent (De Gramont, 1987). Complications more often develop postpartum, and half of affected women develop venous thrombosis during the puerperium (Fieni, 2006; Greene, 1983; Ray, 2000). In one report of 27 pregnancies in 22 women, the maternal mortality rate was 8 percent and related to postpartum thrombosis (de Guibert, 2011).
Severe Preeclampsia and Eclampsia
Fragmentation or microangiopathic hemolysis with thrombocytopenia is relatively common with severe preeclampsia and eclampsia (Kenny, 2014; Pritchard, 1976). Mild degrees are likely present in most cases of severe preeclampsia and may be referred to as HELLP syndrome—hemolysis, elevated liver enzymes, and low platelet count (Chap. 40, p. 742).
Acute Fatty Liver of Pregnancy
This syndrome is associated with moderate to severe hemolytic anemia (Nelson, 2013). It is discussed further in Chapter 55 (p. 1086).
Bacterial Toxins
The most fulminant acquired hemolytic anemia encountered during pregnancy is caused by the exotoxin of Clostridium perfringens or by group A β-hemolytic streptococcus (Chap. 47, p. 949). Endotoxin of gram-negative bacteria, that is, lipopolysaccharide—especially with bacteremia from severe pyelonephritis—may be accompanied by hemolysis and mild to moderate anemia (Cox, 1991).
Inherited Erythrocyte Membrane Defects
The normal erythrocyte is a biconcave disc. Its shape allows numerous cycles of reversible deformations as the erythrocyte withstands arterial shearing forces and negotiates through splenic slits half the width of its cross-sectional diameter. Several genes encode expression of erythrocyte structural membrane proteins or intraerythrocytic enzymes. Various mutations of these genes may result in inherited membrane defects or enzyme deficiencies that destabilize the lipid bilayer. The loss of lipids from the erythrocyte membrane causes a surface area deficiency and poorly deformable cells that undergo hemolysis. Anemia severity depends on the degree of rigidity or decreased distensibility. Erythrocyte morphology similarly is dependent on these factors, and these disorders are usually named after the most dominant red-cell shape characteristic of the disorder. Three examples are hereditary spherocytosis, pyropoikilocytosis, and ovalocytosis.
Hereditary Spherocytosis. Hemolytic anemias that comprise this group of inherited membrane defects are probably the most common identified in pregnant women. Mutations are usually an autosomally dominant, variably penetrant spectrin deficiency. Others are autosomally recessive or de novo gene mutations that result from deficiency of ankyrin, protein 4.2, moderate band 3, or combinations of these (Gallagher, 2010; Yawata, 2000). Clinically, varying degrees of anemia and jaundice result from hemolysis. Diagnosis is confirmed by identification of spherocytes on peripheral smear, reticulocytosis, and increased osmotic fragility (Fig. 56-2).
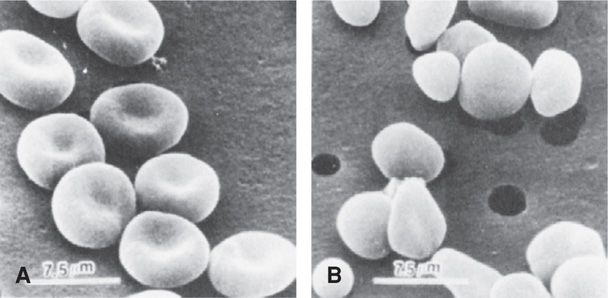
FIGURE 56-2 Scanning electron micrograph showing (A) normal-appearing erythrocytes from a heterozygous carrier of recessive spherocytosis, and (B) from her daughter, a homozygote with severe anemia. (From Agre, 1989, with permission.)
Spherocytic anemias may be associated with the so-called crisis that is characterized by severe anemia from accelerated hemolysis, and it develops in patients with an enlarged spleen. Infection can also accelerate hemolysis or suppress erythropoiesis to worsen anemia. An example of the latter is infection with parvovirus B19 (Chap. 64, p. 1244). In severe cases, splenectomy reduces hemolysis, anemia, and jaundice.
Pregnancy. In general, women with inherited red-cell membrane defects do well during pregnancy. Folic acid supplementation is given to sustain erythropoiesis. Pregnancy outcomes in women with hereditary spherocytosis cared for at Parkland Hospital were reported by Maberry and associates (1992). Twenty-three women with 50 pregnancies were described. In late pregnancy, these women had hematocrits ranging from 23 to 41 volume percent—mean 31. Their reticulocyte count ranged from 1 to 23 percent. Eight women miscarried. Four of 42 infants were born preterm, but none were growth restricted. Infection in four women intensified hemolysis, and three of these required transfusions. Similar results were reported by Pajor and coworkers (1993) in 19 pregnancies in eight women.
Because these disorders are inherited, the newborn may be affected. Celkan and Alhaj (2008) report prenatal diagnosis via cordocentesis at 18 weeks’ gestation and testing for osmotic fragility. Those with hereditary spherocytosis may manifest hyperbilirubinemia and anemia shortly after birth.
Erythrocyte Enzyme Deficiencies
An intraerythrocytic deficiency of enzymes that permit anaerobic glucose metabolism may cause hereditary nonspherocytic anemia. Most of these mutations are autosomal recessive traits, and pyruvate kinase deficiency is probably the most clinically significant. Another is X-linked glucose-6-phosphate dehydrogenase (G6PD) deficiency (Puig, 2013). Other rare enzyme abnormalities may cause varying degrees of chronic hemolysis. As discussed on page 1105, most episodes of severe anemia with enzyme deficiencies are induced by drugs or infections. During pregnancy, oxidant drugs are avoided, infections are treated promptly, and iron and folic acid are given.
Pyruvate kinase deficiency is associated with variable anemia and hypertensive complications (Wax, 2007). Due to recurrent transfusions in homozygous carriers, iron overload is frequent, and associated myocardial dysfunction should be monitored (Dolan, 2002). The fetus that is homozygous for this mutation may develop hydrops fetalis from anemia and heart failure (Chap. 15, p. 315). Gilsanz and colleagues (1993) used funipuncture to diagnose fetal anemia and pyruvate kinase deficiency.
Glucose-6-phosphate dehydrogenase deficiency is complex because there are more than 400 known enzyme variants. The most common are caused by a base substitution that leads to an amino acid replacement and a broad range of phenotypic severity (Beutler, 1991; Puig, 2013)). In the homozygous or A variant, both X chromosomes are affected, and erythrocytes are markedly deficient in G6PD activity. Approximately 2 percent of African American women are affected. The heterozygous variant that is found in 10 to 15 percent of African American women may confer some degree of protection against malaria (Mockenhaupt, 2003). In both instances, random X-chromosome inactivation—lyonization—results in a variable deficiency of enzyme activity.
During pregnancy, infections or drugs can induce hemolysis in both heterozygous and homozygous women, and the severity is related to enzyme activity. Anemia is usually episodic, although some variants induce chronic nonspherocytic hemolysis. Because young erythrocytes contain more enzyme activity than older erythrocytes, in the absence of bone marrow depression, anemia ultimately stabilizes and is corrected soon after the inciting cause is eliminated. Newborn screening for G6PD deficiency is not recommended by the American College of Medical Genetics (2013) as discussed in Chapter 32 (Table 32-3, p. 632).
 Aplastic and Hypoplastic Anemia
Aplastic and Hypoplastic Anemia
Although rarely encountered during pregnancy, aplastic anemia is a grave complication. It is characterized by pancytopenia and markedly hypocellular bone marrow (Young, 2008). There are multiple etiologies, and at least one is linked to autoimmune diseases (Stalder, 2009). The inciting cause can be identified in approximately a third of cases. These include drugs and other chemicals, infection, irradiation, leukemia, immunological disorders, and inherited conditions such as Fanconi anemia and Diamond-Blackfan syndrome (Green, 2009; Lipton, 2009). The functional defect appears to be a marked decrease in committed marrow stem cells.
Hematopoietic stem-cell transplantation is optimal therapy in a young patient (Young, 2008). Immunosuppressive therapy is given. In some nonresponders, eltrombopag has been used successfully (Olnes, 2012). Definitive treatment is bone marrow transplantation, and approximately three fourths of patients have a good response with long-term survival when treated with antithymocyte globulin and cyclosporine (Rosenfeld, 2003). There is a potential for transplantation with umbilical cord blood–derived stem cells (Moise, 2005; Pinto, 2008). Previous blood transfusions and even pregnancy enhance the risk of graft rejection, which is the most common serious complication, causing two thirds of deaths within the first 2 years (Socié, 1999).
Pregnancy
Hypoplastic or aplastic anemia complicating pregnancy is rare. In most cases, the diagnosis precedes conception, or the condition develops during pregnancy as a chance occurrence. That said, there are a few well-documented cases of pregnancy-induced hypoplastic anemia (Bourantas, 1997; Choudhry, 2002). We have cared for a few such women in whom hypoplastic anemia was first identified during a pregnancy. Anemia and other cytopenias improved or remitted following delivery or pregnancy termination. In some cases, recurrence in a subsequent pregnancy was documented.
Diamond-Blackfan anemia is a rare form of pure red-cell hypoplasia, and approximately 40 percent are familial and have autosomal dominant inheritance (Orfali, 2004). There is usually a good response to glucocorticoid therapy. However, continuous treatment is necessary, and most become at least partially transfusion dependent (Vlachos, 2008). In 64 pregnancies complicated by this syndrome, Faivre and associates (2006) reported that two thirds had complications related to placental vascular etiologies that included miscarriage, preeclampsia, preterm birth, stillbirth, or growth-restricted newborn.
Gaucher disease is an autosomally recessive lysosomal enzyme deficiency characterized by deficient acid β-glucosidase activity. It involves multiple systems, including bone marrow. Affected women have anemia and thrombocytopenia that is usually worsened by pregnancy (Granovsky-Grisaru, 1995). Elstein and colleagues (1997) described six pregnant women whose disease improved when they were given alglucerase enzyme replacement. Imiglucerase therapy, which is human recombinant enzyme replacement therapy, has been available since 1994. European guidelines recommend treatment in pregnancy, whereas the Food and Drug Administration states it may be given in pregnancy with “clear indications” (Granovsky-Grisaru, 2011).
The major risks to pregnant woman with hypoplastic anemia are hemorrhage and infection. Management depends on gestational age, disease severity, and whether treatment has been given. Supportive care includes continuous infection surveillance and prompt antimicrobial therapy. Granulocyte transfusions are given only during infections. Red cells are transfused to improve symptomatic anemia and routinely to maintain the hematocrit at or above 20 volume percent. Platelet transfusions may be needed to control hemorrhage. Even when thrombocytopenia is intense, the risk of severe hemorrhage can be minimized by vaginal rather than cesarean delivery. Maternal mortality rates reported since 1960 have averaged nearly 50 percent (Choudhry, 2002). Better outcomes are reported with more recent series (Kwon, 2006).
Pregnancy after Bone Marrow Transplantation
There have been several reports of successful pregnancies in women who have undergone bone marrow transplantation (Borgna-Pignatti, 1996; Eliyahu, 1994). In their review, Sanders and coworkers (1996) reported 72 pregnancies in 41 women who had undergone transplantation. In the 52 pregnancies resulting in a liveborn infant, almost half were complicated by preterm delivery or hypertension. Our experiences with a few of these women indicate that they have normal pregnancy-augmented erythropoiesis and total blood volume expansion.
POLYCYTHEMIAS
 Secondary Polycythemia
Secondary Polycythemia
Excessive erythrocytosis during pregnancy is usually related to chronic hypoxia from maternal congenital cardiac disease or a chronic pulmonary disorder. Unusually heavy cigarette smoking can cause polycythemia. We have encountered otherwise healthy pregnant women who were heavy smokers, had chronic bronchitis, and had hematocrits ranging from 55 to 60 volume percent! Brewer and colleagues (1992) described a woman with persistent erythrocytosis associated with a placental site tumor. If polycythemia is severe, the probability of a successful pregnancy outcome is low.
 Polycythemia Vera
Polycythemia Vera
This is a primary myeloproliferative hemopoietic stem cell disorder characterized by excessive proliferation of erythroid, myeloid, and megakaryocytic precursors. It is uncommon and likely an acquired genetic disorder of stem cells (Spivak, 2008). Virtually all patients have either a JAK2V617F or a JAK2 exon 12 gene mutation (Harrison, 2009). Serum erythropoietin level measurement is helpful to differentiate polycythemia vera—low values—from secondary erythrocytosis—high values. Symptoms are related to increased blood viscosity, and thrombotic complications are common. Fetal loss has been reported to be high in women with polycythemia vera, and pregnancy outcome may be improved with aspirin therapy (Griesshammer, 2006; Robinson, 2005; Tefferi, 2000).
HEMOGLOBINOPATHIES
 Sickle-Cell Hemoglobinopathies
Sickle-Cell Hemoglobinopathies
Hemoglobin A is the most common hemoglobin tetramer and consists of two α- and two β-chains. In contrast, sickle hemoglobin (hemoglobin S) originates from a single β-chain substitution of glutamic acid by valine, which stems from an A-for-T substitution at codon 6 of the β-globin gene. Hemoglobinopathies that can result in clinical features of the sickle-cell syndrome include sickle-cell anemia—Hb SS; sickle cell-hemoglobin C disease—Hb SC; sickle cell-β-thalassemia disease—either Hb S/B0 or Hb S/B+; and sickle-cell E disease—Hb SE (Stuart, 2004). All are also associated with increased maternal and perinatal morbidity and mortality.
Inheritance
Sickle-cell anemia originates from the inheritance of the gene for S hemoglobin from each parent. In the United States, 1 of 12 African Americans has sickle-cell trait, which results from inheritance of one gene for hemoglobin S and one for normal hemoglobin A. The computed incidence of sickle-cell anemia among African Americans is 1 in 576 (1/12 × 1/12 × 1/4 = 1/576). But, the disease is less common in adults and therefore during pregnancy because of earlier mortality, especially during early childhood.
Hemoglobin C originates from a single β-chain substitution of glutamic acid by lysine at codon 6 of the β-globin gene. Approximately 1 in 40 African Americans has the gene for hemoglobin C. Thus, the theoretical incidence for coinheritance of the gene for hemoglobin S and an allelic gene for hemoglobin C in an African American child is about 1 in 2000 (1/12 × 1/40 × 1/4). β-Thalassemia minor is approximately 1 in 40, thus S-β-thalassemia also is found in approximately 1 in 2000 (1/12 × 1/40 × 1/4).
Pathophysiology
Red cells with hemoglobin S undergo sickling when they are deoxygenated, and the hemoglobin aggregates. Constant sickling and unsickling cause membrane damage, and the cell may become irreversibly sickled. Events that slow erythrocyte transit through the microcirculation contribute to vasoocclusion. These include endothelial cell adhesion, erythrocytic dehydration, and vasomotor dysregulation. Clinically, the hallmarks of sickling episodes are periods during which there is ischemia and infarction in various organs. These produce clinical symptoms, predominately pain, which is often severe—the sickle-cell crisis. There may be aplastic, megaloblastic, sequestration, and hemolytic crises.
Chronic and acute changes from sickling include bony abnormalities such as osteonecrosis of femoral and humeral heads, renal medullary damage, autosplenectomy in homozygous SS patients and splenomegaly in other variants, hepatomegaly, ventricular hypertrophy, pulmonary infarctions, pulmonary hypertension, cerebrovascular accidents, leg ulcers, and a propensity for infection and sepsis (Driscoll, 2003; Gladwin, 2004; Stuart, 2004). Of increasing importance is acquisition of pulmonary hypertension, which can be demonstrated in 20 percent of adults with SS hemoglobin (Gladwin, 2008). Depending on its severity, this complication increases the relative risk for death from four- to 11-fold. Another emerging problem with improved survival is chronic sickle-cell disease nephropathy (Maigne, 2010). The median age at death for women is 48 years. Even so, Serjeant and associates (2009) described a cohort of 102 patients followed since birth in which 40 were still alive at 60 to 87 years!
Treatment
Good supportive care is essential to prevent mortality in patients with sickle-cell syndromes. Specific therapies are evolving, and many are still experimental (Stuart, 2004). One treatment is hemoglobin F induction given for both sickling and thalassemia syndromes. These drugs stimulate gamma-chain synthesis. This increases hemoglobin F (fetal hemoglobin), which inhibits hemoglobin S polymerization. One inducing agent, hydroxyurea, when given to patients with moderate to severe disease, increases hemoglobin F production and mitigates erythrocyte membrane damage. This reduces the number of clinical sickling episodes (Platt, 2008). It is not known yet if hydroxyurea increases long-term patient survival (Brawley, 2008). Hydroxyurea is teratogenic in animals, although a preliminary 17-year surveillance of antenatally exposed children was reassuring (Ballas, 2009; Briggs, 2011; Italia, 2010).
Various forms of hemopoietic cell transplantation are emerging as “cures” for sickle-cell syndromes and severe thalassemias (Hsieh, 2009). Oringanje and coworkers (2009) performed a Cochrane Review and found that only observational studies have been reported. Bone marrow transplantation, as discussed on page 1107, has 5-year survival rates that exceed 90 percent (Dalle, 2013). Cord-blood stem-cell transplantation from related donors has also shown great promise (Shenoy, 2013). One intriguing treatment uses cells taken for prenatal diagnosis from a fetus destined to have sickle-cell anemia. Research suggests these cells can be conditioned to produce hemoglobin A and used for replacement after birth (Ye, 2009).
Pregnancy and Sickle-Cell Syndromes
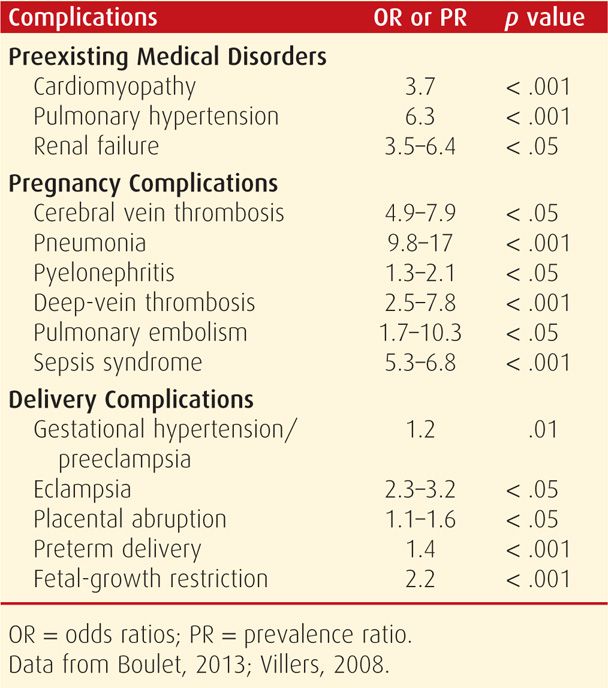
Pregnancy is a serious burden to women with any of the major sickle hemoglobinopathies, particularly those with hemoglobin SS disease. Two large studies define this relationship. The first, by Villers and colleagues (2008), included 17,952 births delivered of women with sickle-cell syndromes from 2000 through 2003. The second study, by Chakravarty and associates (2008), was from 2002 through 2004 and included 4352 pregnancies. A more recent cohort study of 1526 women was reported by Boulet and coworkers (2013). Common obstetrical and medical complications and their relative risks from these studies are shown in Table 56-2. Added to these are findings of Chakravarty and associates (2008), who reported significantly increased risks for renal failure, gestational hypertension, and fetal-growth restriction.
TABLE 56-2. Increased Rates for Maternal Complications in Pregnancies Complicated by Sickle-Cell Syndromes
Maternal morbidity common in pregnancy includes ischemic necrosis of multiple organs, especially bone marrow, that causes episodes of severe pain. Pyelonephritis, pneumonia, and other pulmonary complications are frequent. The acute chest syndrome is manifest by the radiological appearance of a new pulmonary infiltrate accompanied by fever and respiratory symptoms. There are four precipitants of this—infection, marrow emboli, thromboembolism, and atelectasis (Medoff, 2005). Of these, infection causes approximately half of cases and results from typical bacteria and viruses. When the chest syndrome develops, the mean duration of hospitalization is 10.5 days. Mechanical ventilation is required in approximately 15 percent, and the mortality rate is about 3 percent (Gladwin, 2008).
Despite these complications, the maternal mortality rate has decreased. And although improved, perinatal morbidity and mortality rates remain formidable (Yu, 2009). Some perinatal outcomes reported since 2000 are shown in Table 56-3. In addition to increased risks for preterm birth shown in Table 56-2, the frequency of fetal-growth restriction and perinatal mortality are daunting.
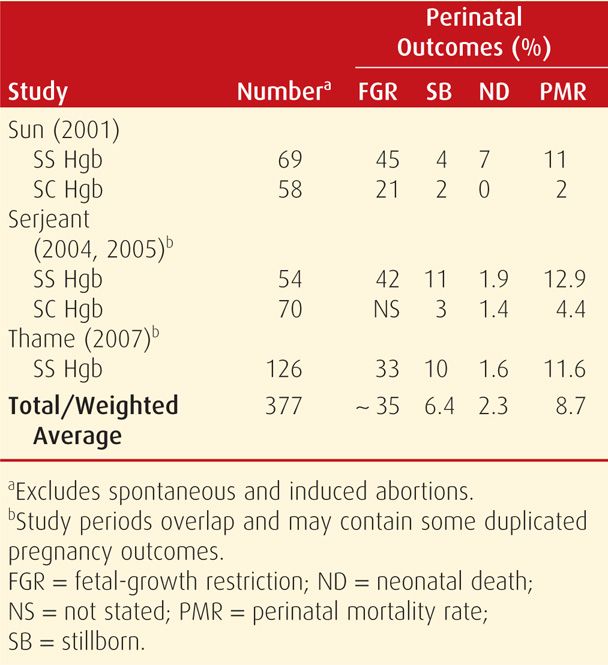
Hemoglobin SC
In nonpregnant women, morbidity and mortality rates from SC disease are appreciably lower than those from sickle-cell anemia. Indeed, fewer than half of women with SC disease have symptoms before pregnancy. In our experiences, affected pregnant and puerperal women suffer attacks of severe bone pain and episodes of pulmonary infarction and embolization—acute chest syndrome—more commonly compared with when they are not pregnant (Cunningham, 1983). It is arguable whether SC disease has a maternal mortality rate equivalent to that of hemoglobin SS disease (Pritchard, 1973; Serjeant, 2005). In the reports shown in Table 56-3, the perinatal mortality rate is somewhat greater than that of the general obstetrical population, but nowhere as great as with sickle-cell anemia (Tita, 2007).
Management During Pregnancy
Women with sickle-cell hemoglobinopathies require close prenatal observation. These women maintain hemoglobin mass by intense hemopoiesis to compensate for the markedly shortened erythrocyte life span. Thus, any factor that impairs erythropoiesis or increases red cell destruction or both aggravates the anemia. Prenatal folic acid supplementation with 4 mg daily is needed to support the rapid red blood cell turnover (American College of Obstetricians and Gynecologists, 2013b).
One danger is that a symptomatic woman may categorically be considered to be suffering from a sickle-cell crisis. As a result, serious obstetrical or medical problems that cause pain, anemia, or both may be overlooked. Some examples are ectopic pregnancy, placental abruption, pyelonephritis, or appendicitis. Thus, the term “sickle-cell crisis” importantly should be applied only after all other possible causes of pain or fever or worsening anemia have been excluded. Pain with sickle-cell syndromes is caused by intense sequestration of sickled erythrocytes and infarction in various organs. These episodes may develop acutely, especially late in pregnancy, during labor and delivery, and early in the puerperium. Because bone marrow is frequently involved, intense bone pain is common.
A system for care of these women has been appropriately stressed by Rees and colleagues (2003). Marti-Carvajal and coworkers (2009) performed a Cochrane review and reported that no randomized trials have evaluated treatment during pregnancy. At minimum, intravenous fluids are given and opioids administered promptly for severe pain. Oxygen via nasal cannula may decrease the intensity of sickling at the capillary level. We have found that red cell transfusions after the onset of severe pain do not dramatically improve the intensity of the current pain crisis and may not shorten its duration. Conversely, as discussed later, prophylactic transfusions almost always prevent further vasoocclusive episodes and pain crises. Recent reports suggest benefits from epidural analgesia used for severe sickle-cell crisis in nonlaboring obstetrical patients (Verstraete, 2012; Winder, 2011). During the past few years, we have encountered several affected women who apparently are habituated to narcotics given chronically to alleviate sickle-cell pain. It is problematic that such women complain of “sickling pain” even when they have effectively undergone exchange transfusion with normal AA-hemoglobin-containing donor red cells.
Rates of covert bacteriuria and acute pyelonephritis are increased substantively, and screening and treatment for bacteriuria are essential. If pyelonephritis develops, sickle cells are extremely susceptible to bacterial endotoxin, which can cause dramatic and rapid red cell destruction while simultaneously suppressing erythropoiesis. Pneumonia, especially due to Streptococcus pneumoniae, is common. The CDC (2013a) recommends the following vaccines for those with sickle-cell disease and all asplenic patients: polyvalent pneumococcal, Haemophilus influenzae type B, and meningococcal vaccines.
Pulmonary complications are often encountered. Acute chest syndrome is characterized by pleuritic chest pain, fever, cough, lung infiltrates, and hypoxia, and usually also by bone and joint pain (Vichinsky, 2000). The spectrum of its pathology includes infection, infarction, pulmonary sequestration, and fat embolization from bone marrow. At least for nonpregnant adults, some recommend rapid simple or exchange transfusions to remove the “trigger” for acute chest syndromes (Gladwin, 2008). In a cohort study of nonpregnant patients, Turner and colleagues (2009) reported that there were no increased benefits of exchange versus simple transfusions, and the former were associated with fourfold increased blood usage. Recurrent episodes of acute chest syndrome may lead to restrictive chronic lung disease or arteriolar vasculopathy and pulmonary hypertension.
Pregnant women with sickle-cell anemia usually have some degree of cardiac dysfunction from ventricular hypertrophy. There is increased preload, decreased afterload, normal ejection fraction, and a high cardiac output. Chronic hypertension worsens this pattern (Gandhi, 2000). During pregnancy, the basal hemodynamic state characterized by high cardiac output and increased blood volume is augmented (Veille, 1994). Although most women tolerate pregnancy without problems, complications such as severe preeclampsia or serious infections may result in ventricular failure (Cunningham, 1986). As discussed in Chapter 49 (p. 986), heart failure caused by pulmonary hypertension must also be considered (Chakravarty, 2008; Stuart, 2004).
In the review of 4352 pregnancies in women with sickle-cell syndromes noted earlier, Chakravarty and associates (2008) reported significantly increased pregnancy complications compared with the total population of 11.2 million women. Compared with controls, women with sickling disorders had a 63-percent rate of nondelivery-related admissions. They had a 1.8-fold increased incidence of hypertensive disorders—19 percent; a 2.9-fold increased rate of fetal-growth restriction—6 percent; and a 1.7-fold increased cesarean delivery rate—45 percent.
Prophylactic Red Cell Transfusions. There are several clinical situations for which prophylactic transfusions have been shown to decrease morbidity from sickle-cell syndromes. For example, chronic transfusion therapy prevents strokes in high-risk children. However, there are no randomized trials of such therapy to prevent pulmonary hypertension and chronic sickle-cell lung disease (Cho, 2014). Preoperative transfusions improved some postoperative outcomes (Howard, 2013). During pregnancy, the most dramatic impact of prophylactic transfusions has been on maternal morbidity. In an observational 10-year prospective study at Parkland Hospital, we offered prophylactic transfusions to all pregnant women with sickle-cell syndromes. Transfusions were given throughout pregnancy to maintain the hematocrit above 25 volume percent and the portion of hemoglobin S below 60 percent (Cunningham, 1979). There was minimal maternal morbidity, and erythropoiesis suppression was not problematic. Their outcomes were compared with historical controls who were not routinely transfused. Morbidity and hospitalizations were significantly reduced in the transfused group (Cunningham, 1983). Others have reported similar data (Grossetti, 2009; Howard, 1995). Still, adverse perinatal outcomes are prevalent (Ngô, 2010).
In a multicenter trial, Koshy and coworkers (1988) randomly assigned 72 pregnant women with sickle-cell syndrome to prophylactic or indicated transfusions. They reported a significant decline in the incidence of painful sickle-cell crises with prophylactic transfusions but no differences in perinatal outcomes. Because of risks inherent with blood administration, they concluded that prophylactic transfusions were not necessary.
There is no doubt that morbidity from multiple transfusions is significant. Up to 10 percent of women had a delayed hemolytic transfusion reaction, and infections are major concerns (Garratty, 1997). From our experiences from Parkland Hospital cited above, we found the incidence of red cell alloimmunization to be 3 percent per unit of blood transfused (Cox, 1988). Similarly, in 12 studies reviewed by Garratty (1997), alloimmunization developed in a fourth of women undergoing chronic transfusions. According to Kacker and colleagues (2014), it not cost effective to extensively match blood donors for these women. Finally, although worrisome, we found no evidence of transfusional iron overload, hemochromatosis, or chronic hepatitis in liver biopsies performed in 40 women transfused during pregnancy (Yeomans, 1990).
Because of what some consider marginal benefits, routine prophylactic transfusions during pregnancy remain controversial (American College of Obstetricians and Gynecologists, 2013b). Similar conclusions were reached after a Cochrane Database analysis (Okusayna, 2013). Current consensus is that their use should be individualized. For example, some clinicians recommend prophylactic transfusions for women with either frequent vasoocclusive episodes or poor obstetrical outcomes (Castro, 2003).
Fetal Assessment. Because of the high incidence of fetal-growth restriction and perinatal mortality, serial fetal assessment is necessary. According to the American College of Obstetricians and Gynecologists (2013b), a plan for serial sonographic examinations and antepartum fetal surveillance is reasonable. Anyaegbunam and colleagues (1991) evaluated fetal well-being during 39 sickling crises in 24 women. Almost 60 percent had nonreactive stress tests, which became reactive with crisis resolution, and all had an increased uterine artery systolic-diastolic (S/D) ratio. At the same time, there were no changes in umbilical artery S/D ratios (Chap. 10, p. 219). These investigators concluded that transient effects of sickle-cell crisis do not compromise umbilical, and hence fetal, blood flow. At Parkland Hospital, we serially assess these women with sonography for fetal growth and amnionic fluid volume changes. Nonstress or contraction stress tests are not done routinely unless complications such as fetal-growth restriction develop or fetal movement is reported to be diminished.
Labor and Delivery. Management is essentially identical to that for women with cardiac disease (Chap. 49, p. 978). Women should be kept comfortable but not oversedated. Labor epidural analgesia is ideal, and conduction analgesia seems preferable for operative delivery (Camous, 2008). Compatible blood should be available. If a difficult vaginal or cesarean delivery is contemplated, and the hematocrit is < 20 volume percent, then packed erythrocyte transfusions are administered. There is no categorical contraindication to vaginal delivery, and cesarean delivery is reserved for obstetrical indications (Rogers, 2010). Circulatory overload and pulmonary edema from ventricular failure should be avoided.
Contraception and Sterilization
Because of chronic debility, complications caused by pregnancy, and the predictably shortened life span of women with sickle-cell anemia, contraception and possibly sterilization are important considerations. Many clinicians do not recommend combined hormonal pills because of potential adverse vascular and thrombotic effects. After a systematic review, however, it was concluded that there was no increase in complications with their use in women with sickle-cell syndromes (Haddad, 2012). The CDC (2013b) regards the contraceptive pill, patch, and ring along with the copper intrauterine device (IUD) as having “advantages that generally outweigh theoretical or proven risks.” All progestin-only methods may be used without restrictions. Because progesterone has been long known to prevent painful sickle-cell crises, low-dose oral progestins or progesterone injections or implants seem ideal. In one study, de Abood and associates (1997) reported significantly fewer and less intense pain crises in women given depot medroxyprogesterone intramuscularly.
Sickle-Cell Trait
The heterozygous inheritance of the gene for hemoglobin S results in sickle-cell trait, or AS hemoglobin. Hemoglobin A is most abundant, and the amount of hemoglobin S averages approximately 30 percent in each red cell. The frequency of sickle-cell trait among African Americans averages 8 percent. There is evidence that carriers have occasional hematuria, renal papillary necrosis, and hyposthenuria (Tsaras, 2009). And although controversial, we believe that sickle-cell trait is not associated with increased rates of abortion, perinatal mortality, low birthweight, or pregnancy-induced hypertension (Pritchard, 1973; Tita, 2007; Tuck, 1983). One unquestioned relationship is the twofold increased incidence of asymptomatic bacteriuria and urinary infection. Sickle-cell trait therefore should not be considered a deterrent to pregnancy on the basis of increased maternal risks.
Preliminary findings from a cohort study by Austin and coworkers (2009) suggested that African American women with sickle trait may have a higher incidence of venous thromboembolism when using hormonal contraceptives compared with women without the trait. However, more data are needed before these effective contraceptive methods are withheld from trait-positive women.
Inheritance is a concern for the infant of a mother with sickle-cell trait whenever the father carries a gene for abnormal hemoglobins that include S, C, and D or for β-thalassemia trait. Prenatal diagnosis through amniocentesis, chorionic villus sampling (CVS), or preimplantation genetic evaluation is available (Chap. 14, p. 297).
 Hemoglobin C and C-β-Thalassemia
Hemoglobin C and C-β-Thalassemia
Approximately 2 percent of African Americans are heterozygous for hemoglobin C, but even if homozygous, hemoglobin C is innocuous (Nagel, 2003). Neither cause severe anemia or adverse pregnancy outcomes. However, when coinherited with sickle-cell trait, hemoglobin SC is the second most common serious sickle-cell syndrome.
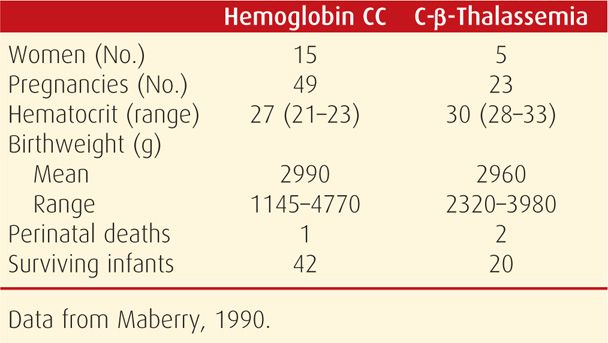
Pregnancy and homozygous hemoglobin CC disease or hemoglobin C-β-thalassemia are relatively benign associations. Maberry and colleagues (1990) reported our experiences from Parkland Hospital as shown in Table 56-4. Other than mild to moderate anemia, pregnancy outcomes were not different compared with those of the general obstetrical population. When severe anemia is identified, iron or folic acid deficiency or some other superimposed cause should be suspected. Supplementation with folic acid and iron is indicated.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


