The Centers for Disease Control and Prevention’s definition of anemia in pregnancy is hemoglobin (Hgb) or hematocrit (Hct) value less than the fifth percentile in a healthy reference population at the same stage of pregnancy.
Typical values include Hgb < 11.0 g/dL in the first and third trimesters and <10.5 in the second trimester (Fig. 20-1).
Racial differences have been noted, with lower Hgb and Hct levels seen in African American women compared with white women. The Institute of Medicine suggests lowering the normal value for Hgb by 0.8 g/dL and Hct by 2% in African Americans.
Anemia is commonly classified by mean corpuscular volume (MCV) as normocytic, microcytic, and macrocytic (Table 20-1).
Iron deficiency anemia is the most common anemia diagnosed during pregnancy, accounting for nearly 50% to 75% of all cases.
Diagnosis is based on insidious symptom onset, such as weakness and lethargy, and in severe cases, glossitis, stomatitis, koilonychia (in which the outer surfaces of the nails are concave), pica, impaired thermogenesis, and gastritis.
Laboratory findings: If a microcytic anemia is present, iron studies are indicated (Table 20-2). Measurement of serum ferritin levels has the greatest sensitivity and specificity for the diagnosis of iron deficiency. Serum ferritin levels <10 to 15 ng/mL (or µ/L) generally indicate iron deficiency anemia. A typical diagnostic cutoff is <12 ng/mL.
Treatment: Although it is important for pregnant women to maintain healthy iron levels, insufficient data exist regarding the benefits of iron supplementation for anemia prophylaxis and treatment during pregnancy. The Centers for Disease Control and Prevention currently recommends daily elemental iron supplementation (30 mg) for prophylaxis and 60 to 120 mg of daily elemental iron if iron deficiency anemia has been diagnosed. A 325-mg tablet of ferrous sulfate contains 65-mg elemental iron; a 300-mg tablet of ferrous gluconate contains 34-mg elemental iron. The American College of Obstetricians and Gynecologists (ACOG)
guidelines recommend supplemental iron for women with iron deficiency anemia. For patients who do not respond to or cannot tolerate oral therapy, or for those with severe anemia, intravenous (IV) iron is an alternative. In women with Hgb <6 g/dL, fetal well-being may be compromised secondary to abnormal fetal oxygenation, and maternal transfusion may be indicated.
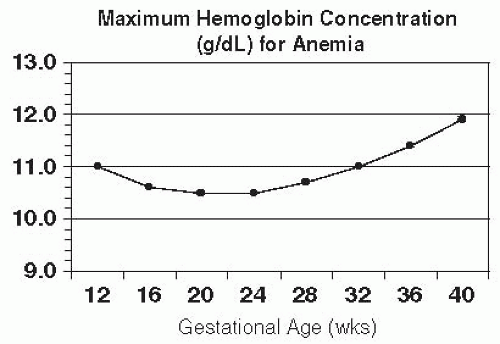 Figure 20-1. Cutoff values for anemia, defined as a hemoglobin level below the fifth percentile based on values from pregnant women with adequate iron supplementation. (Data adapted from Centers for Disease Control and Prevention. Recommendations to prevent and control iron deficiency in the United States. MMWR Recomm Rep 1998;47[RR-3]:1-29.) |
Hemoglobinopathies are genetic abnormalities in the globin portion of the hemoglobin molecule (HbA) that can either be qualitative, resulting in structural abnormalities
like sickle cell anemia, or quantitative, resulting in a decreased number of normal globin chains as in the thalassemias. Normal Hgb is composed of 96% to 97% HbA, 2% to 3% hemoglobin alpha 2 (HbA2), and <1% fetal hemoglobin (HbF).
TABLE 20-1 Classification of Anemia by Mean Corpuscular Volume | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||
TABLE 20-2 Laboratory Studies in Various Anemias | ||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||
Sickle cell disease (SCD) describes a group of hemoglobinopathies that involve sickle hemoglobin (HbS), including SCD (often called “sickle cell anemia”), sickle cell hemoglobin C (HbSC), and sickle-thalassemia hemoglobin (HbS-Thal). Homozygosity for HbS (HbSS) is the most common phenotype, occurring primarily among people from sub-Saharan Africa, South and Central America, Saudi Arabia, India, and Mediterranean countries. Approximately 1 in every 500 to 600 African American newborns has SCD, an autosomal recessive disorder. Affected patients may experience hemolytic anemia, recurrent pain crises, infection, and infarction of more than one organ system. HbS results from a single substitution of valine for glutamic acid at the sixth position of the beta (β)-globin chain. When deoxygenated, HbS is less soluble and tends to polymerize into rigid aggregates and distort the red blood cell into a sickle shape. These cells undergo extravascular hemolysis, leading to a severe chronic anemia, and may become trapped in the microvasculature, causing vascular obstruction, ischemia, and infarction. This cascade results in a vaso-occlusive crisis, which can be associated with severe pain, fever, organ dysfunction, and tissue necrosis. Vaso-occlusive crises may be triggered by infection, hypoxia, acidosis, dehydration, or psychological stress. A serious complication is acute chest syndrome, one of the leading causes of hospitalization and death in patients with SCD. Acute chest syndrome is characterized by a combination of respiratory symptoms, new lung infiltrates, and fever.
Diagnosis: The anemia is normocytic, normochromic with an Hgb concentration of 6 to 10 g/dL and Hct of 18% to 30%. The reticulocyte count is increased to 3% to 15%. Lactate dehydrogenase is elevated, and haptoglobin is decreased. The peripheral blood smear may show sickle cells, target cells, and Howell-Jolly bodies. Diagnosis is confirmed by Hgb electrophoresis, which typically shows 85% to 100% HbS, absent HbA, normal HbA2, and moderately elevated HbF (usually < 15%). Jaundice may result from red blood cell destruction, leading to unconjugated hyperbilirubinemia.
Treatment: Hydroxyurea may be used to reduce intracellular sickling but is not recommended in pregnancy because it is teratogenic in animal studies. Infections are treated aggressively with antibiotics. Severe anemia is treated with blood transfusion. Pain crises are managed with oxygen, hydration, and analgesia. Controversy surrounds prophylactic exchange transfusion and is reserved for the most severe cases. Additionally,
the risks involved with transfusions must be taken into account. Advantages of transfusion are an increase in HbA level, which improves oxygen-carrying capacity and a decrease in HbS-carrying erythrocytes. If a transfusion is given, leukocyte-depleted packed red cells, phenotyped for major and minor antigens, should be used.
Pregnancy considerations: Patients with SCD are at increased risk for sickling during pregnancy because of increased metabolic requirements, vascular stasis, and a relative hypercoagulable state. Complications during pregnancy in women with SCD include an increased risk of spontaneous abortion, intrauterine growth restriction (IUGR), fetal death in utero, low birth weight, preeclampsia, and premature birth. Women with SCD also experience greater risk of urinary tract infection (UTI), bacteriuria, pulmonary infections and infarction, and, possibly, more painful crises. Due to elevated risk of UTI, a urine culture should be evaluated at minimum in every trimester and treated correspondingly. Women with SCD should receive the pneumococcal vaccine before pregnancy and folate supplementation of 1 to 4 mg/day. Iron supplements should be prescribed only if iron is deficient. The intensity of fetal surveillance varies according to the clinical severity of the disease. In severe cases, twice weekly assessment of fetal well-being should begin at 32 weeks’ gestation, and monthly sonography should be performed to evaluate fetal growth. All African American patients should undergo an Hgb electrophoresis to assess carrier status. If both the patient and the father of the baby are found to be hemoglobinopathy carriers, genetic counseling is indicated. Amniocentesis or chorionic villus sampling (CVS) may be offered for prenatal diagnosis. After delivery, patients should practice early ambulation and wear thromboembolic deterrent stockings to prevent thromboembolism.
Regarding contraception, the levonorgestrel-containing intrauterine device (IUD) and progestin-only implants are considered excellent contraceptive options for patients with SCD. No well-controlled studies have evaluated oral contraceptives in SCD; however, low-dose combined contraceptives appear to be a good choice in some women with SCD. The benefits of copper-containing IUDs are debated due to a potential for increased blood loss but copper-containing IUDs are generally considered a safe and effective method of contraception for women with SCD. Progestin-only pills, depot medroxyprogesterone, and barrier devices are also safe for contraception. Medroxyprogesterone acetate (Depo Provera) injections may decrease the number of pain crises.
Sickle cell trait (HbAS) is common in African Americans (1 in 12, or 8%) and is also prevalent in persons of Mediterranean, Middle Eastern, Indian, Caribbean, and Central and South American descents. Women with sickle cell trait have approximately twice the frequency of UTIs compared to the general population, especially during pregnancy, and should be screened each trimester. No direct fetal compromise exists from maternal sickle cell trait. Partners should be screened because the risk of having a child with SCD becomes one in four if the father is also a carrier.
The term thalassemia encompasses a group of inherited blood disorders that can cause severe microcytic hypochromic anemia. Alpha (α)-thalassemia and beta (β)-thalassemia result from absent or decreased production of structurally normal α- and β-globulin chains, respectively, generating an abnormal ratio of α to non-α chains (see Table 20-3). The excess chains form aggregates that lead to ineffective erythropoiesis and/or hemolysis. A broad spectrum of syndromes is possible, ranging from no symptoms to transfusion-dependent anemia and death. Both diseases are transmitted as autosomal recessive traits.
Alpha-thalassemia is associated with Southeast Asian, African, Caribbean, and Mediterranean origin and results from a deletion of one to all α genes, located on chromosome 16. Excess β globins then form β-globin tetramers called HbH. A fetus would be affected because fetal Hgb also requires α chains.
Beta-thalassemia is associated with Mediterranean, Asian, Middle Eastern, Caribbean, and Hispanic origin. More than 200 alterations (mostly point mutations) in β-globin genes, located on chromosome 11, have been reported. The two consequences of these gene defects are the following: β0, which is the complete absence of the β chain, and β+, which is decreased synthesis of the β chain.
Diagnosis: Thalassemia is usually microcytic and hypochromic with an MCV of <80 fL similar to iron deficiency anemia but with important differences in clinical presentation and laboratory testing.
Laboratory findings: In general, thalassemias, especially the traits, are often misdiagnosed as iron deficiency anemia. However, the anemia is not corrected with iron repletion. A microcytic anemia in the absence of iron deficiency suggests thalassemia and additional testing including electrophoresis and iron studies are warranted. Suspicion for the presence of α-thalassemia is raised by the finding of microcytosis and a normal red cell distribution width with minimal or no anemia in the absence of iron deficiency or β-thalassemia. Pedigree studies are often helpful during workup of patients with α-thalassemia. Molecular genetic testing, such as quantitative polymerase chain reaction (PCR), is needed for diagnosis. Quantitative Hgb electrophoresis is required for the diagnosis of β-thalassemia and should be suspected in cases of elevated HbA2 (>3.5%).
Pregnancy and thalassemia
Women with trait status for either thalassemia require no special care.
Women diagnosed with or at high risk for thalassemia should be offered preconception counseling and information about the availability of prenatal diagnosis. First-trimester, DNA-based prenatal testing (CVS) is available if both members of the couple are carriers. Preimplantation genetic diagnosis may also be an option for affected parents.
Women with HbH may have successful pregnancies, with maternal outcome related to the severity of anemia.
Pregnancy may exacerbate the anemia, necessitating transfusions, and place women at an increased risk for preeclampsia, congestive heart failure, and premature delivery.
Information on pregnancy in women with β-thalassemia major or intermedia is more limited, although successful pregnancies have been reported. These women require close medical evaluation and follow-up.
If asplenic, vaccinations for pneumococcus, Haemophilus influenzae, and meningococcus need to be up-to-date.
Thalassemia may confer an increased risk of neural tube defects secondary to folic acid deficiency, so up to 4 mg/day periconceptional folic acid supplementation is recommended. Iron supplements should be prescribed only if iron deficiency is present; otherwise, iron overload can result.
Antepartum fetal testing should be undertaken in anemic thalassemia patients.
Periodic fetal sonography to assess fetal growth as well as nonstress testing to evaluate fetal well-being is recommended.
Ultrasonography is also useful to detect hydrops fetalis but usually at a later gestational age. Options for affected fetuses include intrauterine blood transfusions, which have shown good success in fetuses with hydrops fetalis.
TABLE 20-3 Findings in Thalassemia | |||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


