Chapter 44
Hematologic and Coagulation Disorders
Shiv K. Sharma MD, FRCA, Jill M. Mhyre MD
Chapter Outline
THROMBOCYTOPENIC COAGULOPATHIES
NEURAXIAL ANESTHESIA IN THE PATIENT WITH ONGOING COAGULOPATHY OR PHARMACOLOGIC ANTICOAGULATION
Anemia
Normal Hemoglobin Morphology
Normal adult hemoglobin consists of four polypeptides (two alpha and two beta chains) and the iron-containing prosthetic group (heme or ferriprotoporphyrin IX). In the early embryo, theta (θ) and zeta (ζ) chains are present instead of the alpha (α) chains, and epsilon (ε) chains are present instead of the beta (β) chains. After early embryogenesis, pairs of alpha chains are linked with pairs of either beta, gamma (γ), or delta (δ) chains to form adult hemoglobin (Hgb A = α2β2), fetal hemoglobin (Hgb F = α2γ2), or hemoglobin A2 (Hgb A2 = α2δ2). By term gestation, the ratio of hemoglobin F to hemoglobin A is approximately 1 : 1. By 1 year of age, hemoglobin F typically constitutes less than 1% of total hemoglobin. Although hemoglobin A2 is present, it constitutes less than 2.5% of total adult hemoglobin.
The sequence of amino acids (141 amino acids for alpha chains and 146 for beta chains) defines the primary structure. The three-dimensional shape of each chain defines the secondary structure, and the relationship between the four chains and the heme prosthetic group defines the tertiary structure of the hemoglobin molecule. The binding of the ligands 2,3-diphosphoglycerate (2,3-DPG) and oxygen defines the quaternary structure. The physiology of oxygen transport in the fetus is described in Chapter 5.
Anemia in Pregnancy
During normal pregnancy, plasma volume increases by approximately 50% but red blood cell (RBC) mass increases by only 30%; this differential increase results in the physiologic anemia of pregnancy (see Chapter 2). If the hemoglobin concentration decreases below 10.5 g/dL, the physician should consider other causes of anemia.1,2
Iron deficiency is the most common cause of anemia in pregnancy. It becomes more prevalent as pregnancy advances; in a population-based sample of women in the United States, the prevalence increased from 7% in the first trimester to 14% in the second trimester and 30% in the third trimester of pregnancy.3 Iron-deficiency anemia during the first two trimesters of pregnancy increases the risk for preterm delivery and low birth weight.4–6 In addition to reduced hematocrit, iron-deficiency anemia is characterized by low mean corpuscular volume (MCV) and low total serum iron, ferritin, and transferrin saturation. In the United States, the risk for iron deficiency is increased by advanced parity, short interpregnancy interval, Mexican-American ethnicity, and African race.3,7 Daily oral iron treatment in pregnancy reduces the risk for anemia and low birth weight; a 2013 meta-analysis of randomized controlled trials suggests that there is a clear dose-response relationship for up to a total iron dose of 66 mg/day.8 Doses greater than 60 mg/day increase side effects, including nausea, vomiting, constipation, and abdominal cramps.9 Observational data suggest that oral iron therapy can reduce the incidence of preterm birth.10 Although a 2013 meta-analysis of randomized controlled trials of iron supplementation failed to identify an effect on the incidence of preterm birth or small-for-gestational-age infants, the relative risk confidence intervals were wide.8 Antepartum anemia is a leading risk factor for postpartum blood transfusion11; however, no study has evaluated the impact of antepartum iron supplementation on the risk of postpartum maternal blood transfusion.
Parenteral (intramuscular or intravenous) iron enhances hematologic response compared with oral iron, but formulations that contain dextran may increase risk for venous thrombosis and allergic reactions.9
An elevated hemoglobin concentration (≥ 14.5 g/dL) may reflect inadequate volume expansion and has been associated with adverse pregnancy outcomes, including preterm delivery, small-for-gestational-age infants, and stillbirth.6,12
Thalassemia
The thalassemias are a group of microcytic, hemolytic anemias that result from the reduced synthesis of one or more of the polypeptide globin chains.13 This reduced synthesis leads to (1) an imbalance in globin chain ratios, (2) defective hemoglobin, and (3) erythrocyte damage resulting from excess globin subunits. In α-thalassemia, alpha-chain production is reduced, and in β-thalassemia, beta-chain production is reduced.
α-Thalassemia
There are two alpha-chain loci on each chromosome 16; therefore, there are four genes that can produce alpha chains.14 Because deletions or mutations can affect any or all of these genes, four types of α-thalassemia exist: (1) silent carrier (three functioning genes), (2) α-thalassemia trait (two functioning genes), (3) hemoglobin H disease (one functioning gene), and (4) α0-thalassemia or Bart’s hydrops (no functioning genes). As the number of functioning genes decreases from three to zero, the ratio of alpha to beta chains decreases from 0.8 : 1 to 0.6 : 1 to 0.3 : 1 to 0 : 1. The mRNA production from the second alpha gene exceeds that of the first alpha gene by a factor of 1.5 to 3.14,15 Therefore, deletions of the second alpha gene may produce greater clinical effect. As beta (or beta-like) chains accumulate, they can form tetramers in utero (hemoglobin Bart’s = γ4) or after delivery (hemoglobin H = β4) and appear as Heinz bodies on the peripheral blood smear.
In the United States, 25% to 30% of black women are silent carriers and have slightly smaller (78 to 85 fL) mean corpuscular volume (MCV) than women without thalassemia.16,17 A chromosome lacking one alpha gene is common in Africa, the Mediterranean basin, the Middle East, India, Southeast Asia, Indonesia, and the South Pacific Islands.18 Silent carriers are not at increased risk for adverse outcome during pregnancy or surgery.
The α-thalassemia trait affects 2% to 3% of black women in the United States16,17 and is almost exclusively due to homozygous α+-thalassemia, in which one functional α-globin gene is preserved on each chromosome (α−/α−). These women have an MCV of 70 to 75 fL and mild anemia. They typically are asymptomatic and, beyond the effects of mild anemia, experience no additional risk for adverse outcomes during pregnancy or surgery. Heterozygous α0-thalassemia trait (−−/αα) is common among individuals of Southeast Asian descent. It is phenotypically indistinguishable from homozygous α+-thalassemia trait but introduces the risk for bearing an offspring with hemoglobin H disease or Bart’s hydrops.
Patients with hemoglobin H disease experience moderately severe microcytic anemia, splenomegaly, fatigue, and generalized discomfort. Hemoglobin H (β4) constitutes 2% to 15% of the total hemoglobin in these patients. Affected patients generally do not have a decreased life span, and hospitalization for the treatment of their anemia rarely is required. However, disease severity and prognosis vary, depending on the specific mutations present19; some patients require lifelong transfusion and chelation therapy.
Hemoglobin Barts, or α0-thalassemia, is generally incompatible with life. The disease is found predominantly in Southeast Asia, China, and the Philippines. Affected individuals die in utero or shortly after birth of hydrops fetalis; mothers carrying these fetuses are prone to develop hypertension or peripartum hemorrhage or both.20 Intact neonatal survival has been reported with intrauterine transfusion therapy and postnatal hematopoietic stem cell transplantation.21,22 Antenatal screening for the disease is possible (see later discussion).
β-Thalassemias
In β-thalassemia, the production of beta chains is reduced. There are more than 200 genetic causes for ineffective beta-chain production, including gene deletion, transcription mutations, and RNA-processing mutations.13 Unlike the alpha chains, which have four genes (two on each chromosome 16), beta chains have only one gene on each chromosome 11. Production of mRNA from the second beta-like gene (i.e., delta) is almost completely suppressed. Therefore, there are only two primary forms of β-thalassemia: (1) β0-thalassemia, in which there is no beta-chain formation, and (2) β+-thalassemia, in which some beta-chain production exists. β0-Thalassemia also is called β-thalassemia major or Cooley’s anemia. Individuals who receive β-thalassemia genes from both parents but with mutations of different types often develop a milder form of the disease and require fewer or no transfusions. This condition is known as thalassemia intermedia. Finally, β-thalassemia minor refers to the heterozygous carrier of β-thalassemia.
β-Thalassemia is found most often in persons from the Mediterranean basin, the Middle East, India, Pakistan, and Southeast Asia and less often among persons from Tajikistan, Turkmenistan, Kyrgyzstan, China, and Africa.13
Individuals with β-thalassemia have a relative excess of alpha chains. Excess alpha chains precipitate and form inclusion bodies in red blood cell (RBC) precursors, resulting in anemia secondary to ineffective erythropoiesis and splenic hemolysis.13 In the fetus, the gamma chain is unaffected; therefore, anemia only develops as gamma-chain production ceases during the first year of life.13 In some patients, gamma-chain production continues to a variable extent.13 Thus, the ongoing production of hemoglobin F (even in adults) may minimize the effects of decreased beta-chain production.13
β-Thalassemia Major.
In patients with β-thalassemia major, progressively severe anemia develops beginning in the first few months of extrauterine life.13 The anemia results in tissue hypoxia, increased intestinal absorption of iron, and increased erythropoietin production. The resulting expansion of marrow cavities causes skeletal abnormalities and pathologic fractures. Splenomegaly leads to thrombocytopenia and leukopenia.
RBC transfusions are required to maintain life, and the resulting iron load leads to iron accumulation, first in Kupffer’s cells (noncirculating macrophages found in the liver), then in liver parenchymal cells, and finally in endocrine and myocardial cells. Deposition of iron in endocrine tissues may result in diabetes mellitus, adrenal insufficiency, and infertility.23 Myocardial accumulation of iron can lead to conduction abnormalities and intractable heart failure, which are exacerbated by anemia-induced tachycardia. Heart failure and infection are the most common causes of death.
Patients with β-thalassemia major who present when younger than 2 years of age often have hepatomegaly and a hemoglobin concentration as low as 2 g/dL. Patients who present later in life (2 to 12 years of age) typically have a hemoglobin concentration between 4 and 10 g/dL, with marked anisopoikilocytosis and numerous target cells, nucleated RBCs, and inclusion bodies. Levels of hemoglobin F range from 10% to 90% of the total hemoglobin, and hemoglobin A2 constitutes the remainder of the hemoglobin present.
Treatment includes (1) lifelong transfusion of leukocyte-poor RBCs every 2 to 3 weeks to maintain a hemoglobin concentration greater than 10 g/dL, thus preventing endogenous erythropoiesis; (2) splenectomy; and (3) iron chelation therapy to prevent hemosiderosis.24 Deferoxamine was the first available chelation agent. It has a long record of successful use, but it requires continuous subcutaneous infusion or intermittent intramuscular injection.25,26 Deferiprone and deferasirox are alternative oral chelation drugs; deferiprone has emerged as a superior agent for reducing cardiac iron levels and preventing cardiac morbidity and mortality.25,27 Hematopoietic stem cell transplantation may be curative if a human leukocyte antigen (HLA)-matched family donor without β-thalassemia major is found.24,28 Research exploring the potential of gene therapy is underway.29
It is unusual for patients with β-thalassemia major to become pregnant; nonetheless, transfusion and chelation regimens improve fertility, and assisted reproductive technologies facilitate conception in women with hemosiderosis-related infertility.30 The metabolic demands of pregnancy increase transfusion requirements. Mordel et al.31 reviewed reports of these patients and suggested that up to 8 L of transfused RBCs may be required to maintain the hemoglobin concentration above 10 g/dL during pregnancy.31 It is unclear whether iron-chelation therapy should be continued throughout pregnancy; insufficient evidence is available to refute theoretical concerns about teratogenicity and fetal iron depletion.32,33 Monitoring for maternal cardiac iron deposition and heart failure may be accomplished using modified magnetic resonance imaging and echocardiography, respectively.25,34 Historically, these patients had an increased incidence of spontaneous abortion, intrauterine fetal death, and fetal growth restriction (also known as intrauterine growth restriction).31 Recent case series suggest that among women with normal cardiovascular function, careful transfusion therapy and multidisciplinary care may facilitate uneventful pregnancy.30,35,36
A trial of labor is appropriate, and operative delivery should be reserved for obstetric indications.37 Chronic transfusions increase the risk for alloimmunization, which prolongs the time required to identify compatible allogeneic blood products in the event of peripartum hemorrhage. Intraoperative blood salvage has been safely performed during cesarean delivery in a parturient with thalassemia.38 Postpartum pharmacologic thromboprophylaxis is indicated.37
Extramedullary hematopoiesis can result in vertebral cortical weakening, pathologic fractures, and, rarely, paraplegia. However, in the absence of a major pathologic process of the spine, neuraxial anesthesia can be safely administered.39 Patients with splenomegaly may develop thrombocytopenia; therefore, anesthesia providers should exclude a history of spontaneous hemorrhage and determine the platelet count before initiating a neuraxial procedure.
β-Thalassemia Minor.
The clinical course is usually benign in patients with β-thalassemia minor. The anemia is typically mild (hemoglobin concentration of 9 to 11 g/dL) and is characterized by microcytosis and hypochromatosis. Levels of hemoglobin F range from 1% to 3%, and levels of hemoglobin A2 range from 3.5% to 7%.
Moderate anemia develops only during periods of stress, such as pregnancy and severe infection. Nonetheless, most patients with β-thalassemia minor tolerate pregnancy well, although the incidence of oligohydramnios and fetal growth restriction are greater than in nonthalassemic women.40 Because of an increased rate of RBC turnover and an increased risk for neural tube defects, high-dose folate supplementation is recommended in the first trimester. Transfusions are reserved for patients with hemorrhage or a hemoglobin concentration below 8 g/dL. Infection, which can cause bone marrow suppression, must be treated promptly. β-Thalassemia minor typically does not affect anesthetic management during labor or cesarean delivery.
Antenatal Thalassemia Screening
Among populations at risk for α- or β-thalassemia, antenatal screening can identify couples at increased risk for offspring with a serious hemoglobinopathy. Low maternal and paternal MCV (≤ 80 fL) or mean corpuscular hemoglobin (MCH ≤ 27 pg) with normal serum iron and ferritin should prompt peripheral smear analysis for inclusion bodies or hemoglobin electrophoresis or both.18 The latter test may reveal elevated hemoglobin A2 or hemoglobin F, suggesting β-thalassemia or another hemoglobinopathy (sickle cell trait [AS], sickle cell anemia [SS], or hemoglobin C trait [SC]). α-Thalassemia requires α-globin gene testing for diagnosis because hemoglobin electrophoresis will not detect it.18 Counseling for fetal genetic testing should be offered if both parents carry at least one abnormal hemoglobin gene.18
Prenatal diagnosis can be accomplished with the use of fetal cells obtained by means of chorionic villus sampling or amniocentesis and subjected to DNA analysis.18,37 In the future, cell-free fetal DNA obtained from maternal plasma may provide an alternative source of material for fetal genetic analysis.
Sickle Cell Disease
More than 1000 abnormal α-, β-, γ-, and δ-globin chains have been identified.41 Structural hemoglobinopathies result when these abnormal chains are used to form hemoglobin molecules. The most common abnormal hemoglobins are hemoglobin S, hemoglobin C, hemoglobin D, and hemoglobin E.41 Patients can be homozygous for an abnormal hemoglobin (e.g., hemoglobin SS or sickle cell anemia), heterozygous for an abnormal hemoglobin (e.g., hemoglobin SA or sickle cell trait), or doubly heterozygous for an abnormal hemoglobin (e.g., hemoglobin SC or sickle cell hemoglobin C disease).18,41 The heterozygous state for both the thalassemias and the structural hemoglobinopathies appears to protect against malaria, which may explain their geographic distribution and continued presence in the gene pool.42
A sickle cell disorder refers to a state in which erythrocytes undergo sickling when they are deoxygenated.42 Normal erythrocytes have a biconcave shape. Sickle cells are elongated and crescent shaped, with two pointed ends. Sickle cell disease refers to disorders in which sickling results in clinical signs and symptoms; it includes hemoglobin SS disease, hemoglobin SC disease, hemoglobin SD disease, and sickle cell β-thalassemia.18,42
Sickle Cell Anemia
Epidemiology.
Table 44-1 lists the prevalence of sickle cell anemia and the other common hemoglobinopathies in the adult black population in the United States. The current number of individuals with sickle cell disease in the United States may approach 90,000, with 10% of Hispanic origin43; however, high-quality surveillance data are not available.44
TABLE 44-1
Prevalence of Hemoglobinopathies in the United States in Persons of African Descent
| Type | Estimated Prevalence |
| Traits | |
| Hemoglobin AS | 1 : 12.5 |
| Hemoglobin AC | 1 : 33 |
| β-Thalassemia minor | 1 : 67 |
| Persistent hemoglobin F | 1 : 1000 |
| Sickling Disorders | |
| Hemoglobin SS | 1 : 625 |
| Hemoglobin SC | 1 : 833 |
| Hemoglobin S–β-thalassemia | 1 : 1667 |
| Hemoglobin S–persistent hemoglobin F | 1 : 25,000 |
| Hemoglobin CC | 1 : 4444 |
| β-Thalassemia major | 1 : 17,778 |
| Hemoglobin C–β-thalassemia | 1 : 4444 |
Modified from Motulsky AG. Frequency of sickling disorders in U.S. blacks. N Engl J Med 1973; 288:31-3.
Pathophysiology.
In hemoglobin S molecules, valine is substituted for glutamic acid as the sixth amino acid in the beta chains.41 This substitution results in a propensity for hemoglobin molecules to aggregate when the hemoglobin is in the deoxygenated state. The hemoglobin molecules stack on top of one another and form microtubules.
Oxygen tension is the most important determinant in sickling; other factors that affect sickling are listed in Box 44-1. Hemoglobin S begins to aggregate at a PO2 of less than 50 mm Hg, and all of the hemoglobin S is aggregated at a PO2 of approximately 23 mm Hg. The formation of hemoglobin S aggregates is time dependent45; the proportion of sickled hemoglobin increases with decreasing cardiac output and prolonged venous transit time. If an erythrocyte sickles, it can return to its normal shape once the hemoglobin S becomes oxygenated.41,45 However, repeated sickling cycles produce erythrocyte metabolic abnormalities and membrane damage, eventually leading to irreversible sickling regardless of oxygen tension.41,45 Sickled cells are cleared rapidly from the circulation by the reticuloendothelial system; as a result, the erythrocyte life span is reduced to approximately 12 days.41,45
Sickled cells can form aggregates and lead to vaso-occlusive crises and end-organ injury. Repeated cycles of sickling, vaso-occlusion, reperfusion injury, and acute inflammation can lead to chronic inflammation and inflammatory vascular disease. Elevated levels of cell-free hemoglobin deplete nitric oxide, activate the endothelium, and further exacerbate inflammation.41,42 The reduced erythrocyte life span results in anemia, jaundice, cholecystitis, and a hyperdynamic hemodynamic state.
Marked ventricular hypertrophy can occur in pregnant women with sickle cell disease secondary to increased cardiac output. This may lead to a decrease in ventricular compliance and a deterioration in ventricular diastolic function.46 Anemia also leads to erythroblastic hyperplasia, expansion of medullary spaces, and a loss of cortex in long bones, vertebral bodies, and the skull.41 Vaso-occlusive events can give rise to infarctive crises (which most often occur in the chest, abdomen, back, and long bones), cerebrovascular accidents, and rarely peripheral neuropathy.47 Aggregate formation in the spleen can result in microinfarcts.
Functional asplenia and abnormal neutrophil responses both contribute to susceptibility to infection. Consequently, the incidence of pneumonia and pyelonephritis is higher in pregnant patients with sickle cell disease than healthy pregnant patients. Aplastic crises can occur from depression of erythropoiesis secondary to infection (especially parvovirus) or from marrow failure secondary to folate deficiency during pregnancy.41 During an aplastic crisis, the hemoglobin concentration can decrease rapidly, leading to high-output cardiac failure and death. Sequestration crises can result from the massive pooling of erythrocytes, especially in the spleen. This event occurs more frequently in patients with hemoglobin SC disease or sickle cell β-thalassemia than in patients with other forms of sickle cell disease. In general, a major sequestration crisis is one in which the hemoglobin concentration is less than 6 g/dL and has decreased more than 3 g/dL from the baseline measurement.41
The long-term clinical course of sickle cell disease is highly variable. Higher fetal hemoglobin expression and coincident α-thalassemia were among the first genetic modulators described.48 Subsequent work has identified a complex network of single nucleotide polymorphisms associated with specific complications of sickle cell disease, most prominently the transforming growth factor-beta (TGF-β) family of membrane-bound receptors. These receptors play a role in fibrosis, cell proliferation, hematopoiesis, osteogenesis, angiogenesis, nephropathy, wound healing, and immune response.48
Diagnosis.
In the adult, sickle cell anemia is characterized by (1) a hemoglobin concentration of 6 to 8 g/dL, (2) an elevated reticulocyte count, and (3) the presence of sickle cells on a peripheral blood smear. The diagnosis is confirmed by electrophoresis, thin layer isoelectric focusing, or high-pressure liquid chromatography.41 Because most hemoglobinopathies are inherited as autosomal recessive conditions, prenatal screening for abnormal hemoglobin is recommended in couples at high risk for sickle cell disease.18 In utero, the diagnosis can be made through the use of restriction endonucleases specific for the sickle mutation applied to fetal cells obtained during amniocentesis or chorionic villus sampling.
Interaction with Pregnancy.
Pregnancy typically exacerbates the complications of sickle cell anemia. Maternal mortality from sickle cell disease comprises as many as 1% of all maternal deaths in the United States.49 Thromboembolic complications, infection, cardiomyopathy, and pulmonary hypertension are the most serious maternal medical complications.49 Patients with sickle cell anemia have an increased incidence of preterm labor, placental abruption, fetal growth restriction, hypertension, and eclampsia.49 Intensive fetal surveillance may reduce the risk for intrauterine fetal death.49
Medical Management.
Sickle cell anemia is a chronic anemia; blood transfusions are given only when they are specifically indicated (e.g., acute anemia, aplastic crisis, pneumonia with hypoxemia, before or during surgery).41 The goals of transfusion are to achieve a hemoglobin concentration greater than 8 g/dL and to ensure that hemoglobin A represents more than 40% of the total hemoglobin present. Prophylactic blood transfusions do not appear to alter fetal or maternal mortality, although the frequency of maternal pain crises may be reduced.50,51 If the patient’s baseline hemoglobin concentration is less than 6 to 7 g/dL, simple transfusions with buffy-coat-poor, hemoglobin S–free, washed RBCs should be adequate to meet treatment goals. Otherwise, partial exchange transfusions may be necessary.
Hemoglobin F does not form aggregates with hemoglobin S. Administration of hydroxyurea may enhance the production of hemoglobin F, which may decrease the morbidity and mortality of sickle cell anemia. It is unclear whether hydroxyurea is safe in pregnancy; the drug is known to be carcinogenic, mutagenic, and teratogenic in animals.52 However, among a small series of pregnancies conceived at the time of maternal or paternal hydroxyurea administration, there was no evidence of abnormal pregnancy outcomes or teratogenicity among surviving offspring.52 Bone marrow transplantation is a potentially curative therapy for individuals with complicated sickle cell disease, although HLA-matched donors can be difficult to locate and the procedure is associated with significant morbidity and mortality.28,53
Obstetric Management.
During prenatal visits, the obstetrician should monitor maternal weight gain, blood pressure, urine protein content, and uterine and fetal growth. Antepartum fetal surveillance begins at the time of extrauterine viability. Blood transfusions are reserved for specific indications. There is no evidence that preoperative blood transfusion to achieve a hemoglobin concentration of 10 g/dL improves perioperative outcomes for nonobstetric sickle cell patients, but no trial has evaluated prophylactic blood transfusion before cesarean delivery.42,54 Early preparation of crossmatched blood products should be considered because alloimmunization, and the antigen crossmatching procedures recommended to prevent its development, can prolong crossmatching procedures.42 Finally, postpartum pharmacologic thromboprophylaxis is indicated.55
Anesthetic Management.
Preoperative evaluation should focus on recent sickle cell disease exacerbations, the degree of anemia, and chronic end-organ injury.42 Pulmonary hypertension and high-output heart failure should be excluded with echocardiography.55 Pain control during labor is essential; continuous neuraxial analgesia is recommended.42 Although general anesthesia for cesarean delivery has been associated with postoperative sickling complications, either neuraxial or general anesthesia is acceptable, and the choice of anesthetic technique ultimately depends on the time available to induce anesthesia and the patient’s preference and physical status.56 Principles of anesthetic management include (1) use of crystalloid to maintain intravascular volume, (2) transfusion of RBCs to maintain oxygen-carrying capacity, (3) administration of supplemental oxygen, (4) maintenance of normothermia, (5) prevention of peripheral venous stasis, and (6) provision of appropriate venous thromboembolism prophylaxis.42,55,56
Sickle Cell Disease Variants
If a patient carries one sickle cell gene and another gene for a hemoglobin that has a propensity to sickle, that patient is considered to have sickle cell disease. Patients with hemoglobin SD disease tend to have the mildest form, and patients with SC disease or sickle cell β-thalassemia tend to have more severe disease.41,57
As with the hemoglobin S gene, hemoglobin C is most prevalent among persons of West African descent, whereas hemoglobin D is more often found among persons of African, northern European, and Indian descent, and hemoglobin E is most prevalent among persons of Southeast Asian descent.41 Patients with hemoglobin SC and hemoglobin SD disease tend to be asymptomatic during childhood with only mild anemia. Typically, these individuals do not develop symptoms until the second half of pregnancy. During late pregnancy they may have severe anemia (secondary to splenic sequestration) and splenomegaly. Patients with hemoglobin SC disease also have a tendency to develop bone marrow necrosis, which predisposes to fat emboli. The other clinical manifestations are similar to those observed in patients with sickle cell anemia.
Blood transfusion is recommended only when the hemoglobin concentration is less than 7 to 8 g/dL. Obstetric and anesthetic management are similar to the management of patients with sickle cell anemia.
Patients who are homozygous for hemoglobin C, D, or E typically have mild anemia. Target cells often are observed and splenomegaly is common. Patients who are heterozygous (i.e., one gene for hemoglobin C, D, or E and one gene for normal hemoglobin) are asymptomatic. The diagnosis is confirmed with electrophoresis, thin-layer isoelectric focusing, or high-pressure liquid chromatography.41 Pregnancy typically is well tolerated, and no specific change in obstetric or anesthetic management is required.
Sickle Cell Trait
Sickle cell trait is the most benign form of the sickle cell disorders. It occurs in approximately 8% of black women in the United States. The RBCs of patients with sickle cell trait do not sickle until the PO2 decreases below 15 mm Hg; therefore, RBC life span is normal.58 A study of 65,000 patients with sickle cell trait found only a slight increase in the incidence of renal (hematuria) and pulmonary (emboli) complications compared with patients without sickle cell trait.59 Patients with sickle cell trait are not at increased risk for adverse outcome during surgery.
Autoimmune Hemolytic Anemia
Patients with autoimmune hemolytic anemia produce antibodies to their own RBCs, resulting in hemolysis and varying degrees of anemia. The annual incidence of new cases of autoimmune hemolytic anemia is approximately 1 per 80,000 persons, but the prevalence approaches 1 in 5,000.60 Table 44-2 lists the characteristics of the four main types of autoimmune hemolytic anemia.61 Warm antibodies react with RBCs at a temperature of 35° to 40° C, whereas cold antibodies react optimally at a temperature lower than 30° C. Table 44-3 lists the various causes of autoimmune hemolytic anemia.62
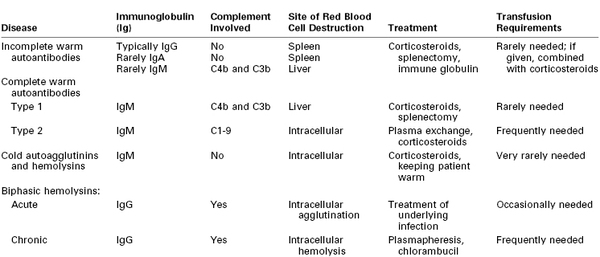
TABLE 44-3
Etiology of Autoimmune Hemolytic Anemias
| Etiology | Approximate Percentage |
| Primary or idiopathic | 43 |
| Secondary: | |
| Neoplasms | 22 |
| Drug-related | 15 |
| Infections | 8 |
| Connective tissue diseases | 5 |
| Other diseases | 5 |
| Pregnancy | 2 |
Patients with warm-reacting antibodies typically respond to treatment with corticosteroids; splenectomy and the anti-CD20 antibody rituximab are second-line therapies.60 After splenectomy, relapses and blood transfusions may lead to the continued requirement for intermittent or continuous corticosteroid therapy. The requirement for extended phenotyping may delay matched blood product availability. In acute hemorrhage, the rapid transfusion of body-temperature, ABO-compatible and Rh-negative blood can be lifesaving, with the expectation that the half-life of transfused red blood cells will be shortened; this practice will allow time to procure compatible blood products.60
In patients with cold-reacting antibodies, the anemia typically is mild and maintenance of normal body and ambient temperatures typically is all that is required to prevent hemolysis.61
Coagulation
Thrombotic and Thrombolytic Pathways
Hemostasis depends on the normal function of vascular tissue, platelets, and coagulation factors. During the initial response to loss of vessel integrity, platelets adhere to exposed collagen, facilitated by the von Willebrand factor (vWF) (primary hemostasis). Platelet activation results in the release of substances that constrict the injured vessels and cause other platelets to adhere and form a hemostatic plug. The platelet plug is not stable, and initiation of the coagulation cascade, followed by deposition and stabilization of fibrin, is necessary for definitive hemostasis (secondary hemostasis). Most coagulation factors circulate in the blood as zymogens, which are converted to active enzymes that in turn convert other zymogens to active enzymes. For example, factor X (a zymogen) is converted to factor Xa (an enzyme), which converts prothrombin (factor II) to thrombin (factor IIa).
In its original conception, the coagulation cascade (Figure 44-1) was believed to propagate within plasma. Subsequent work has located the enzymatic reactions of the extrinsic system primarily to the surface of subendothelial cells and those of the intrinsic system to the activated platelet surface.63 Currently, a widely used model divides the coagulation cascade into three phases: (1) an initiation phase (classical extrinsic pathway), in which small amounts of active coagulation factors are generated; (2) an amplification phase, in which the level of active coagulation factors is boosted; and (3) a propagation phase, in which coagulation factors bind to the membrane of activated platelets, leading to the formation of fibrin clots.63
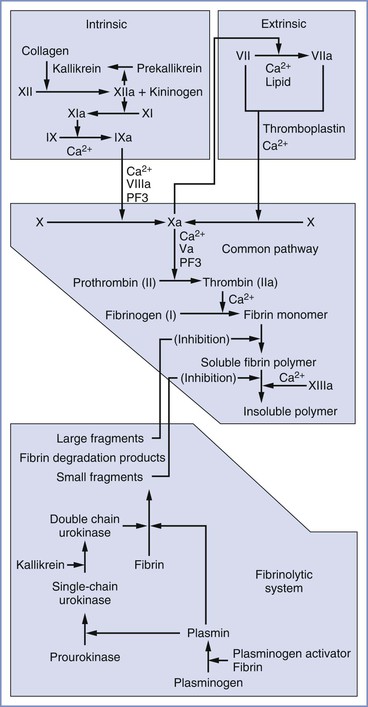
FIGURE 44-1 Components of the extrinsic, intrinsic, and common pathways and the fibrinolytic system. The term cascade is a misnomer that stems from the presence of positive and negative feedback loops in both the coagulation and fibrinolytic systems. PF3, Platelet factor 3.
In the classical extrinsic pathway, tissue damage activates tissue factor (TF) (also known as factor III or thromboplastin) on the surface of extravascular cells (e.g., fibroblasts, smooth muscle cells), which are exposed to the bloodstream after tissue damage. TF has also been identified on the surfaces of syncytiotrophoblasts,64 adhered leukocytes, circulating monocytes, and circulating microparticle membrane vesicles released by inflammatory and tumor cells.63 TF binds factor VII and promotes proteolysis and activation to factor VIIa. On the membrane surface, the TF/VIIa complex converts factor X to Xa and small amounts of factor IX to IXa. Factor Xa amplifies conversion of factor VII to VIIa in the first of many positive feedback loops, and factor Xa forms a complex with factor Va. The membrane-bound prothrombinase complex (i.e., Xa/Va) converts small amounts of soluble prothrombin to thrombin. This thrombin diffuses to the activated platelet surface, where it amplifies the intrinsic coagulation pathway.
In the intrinsic pathway, factor XII binds to a negatively charged substrate (e.g., collagen, platelet phosphatidylserine) and may undergo autolysis to form factor XIIa, or it may be converted to XIIa by trace amounts of XIIa. In addition to activating its own zymogen, factor XIIa converts prekallikrein to kallikrein and factor XI to XIa. High-molecular-weight kininogen can bind factor XI and facilitate its conversion to XIa by XIIa. Kallikrein and high-molecular-weight kininogen also can convert factor XII to XIIa. Factor XIa converts factor IX to IXa, which, with factor VIIIa, converts factor X to Xa. Factor Xa promotes platelet aggregation, and it converts factors V and VIII to factors Va and VIIIa, respectively. Factor Xa, combined with factor Va, converts factor II (prothrombin) to factor IIa (thrombin), a process termed the thrombin burst. Activated platelets provide the primary surface for conversion of factor X to Xa and prothrombin to thrombin. Thrombin converts factors I (fibrinogen), V, VIII, and XIII to factors Ia (fibrin), Va, VIIIa, and XIIIa, respectively. Thrombin also causes platelet activation. Factor XIIIa is required to cross link fibrin strands, which helps form a stable clot.
Clot formation is limited by the natural anticoagulants antithrombin III, proteins C and S, and tissue factor pathway inhibitor (TFPI). Antithrombin III, whose activity is enhanced by heparin, inhibits factor IXa, factor Xa, and thrombin. Protein C is activated by a thrombin-thrombomodulin complex. With protein S as a cofactor, protein C breaks down factors Va and VIIIa. TFPI is produced by endothelial cells and inhibits coagulation by simultaneously binding factor Xa and the TF/factor VIIa complex.
The final component of the coagulation system is the fibrinolytic system, in which plasmin breaks down fibrin. Tissue-type plasminogen activator (t-PA) circulates as an active protease; however, its activity increases dramatically when it binds to fibrin, at which time it converts plasminogen to plasmin. Urokinase-like plasminogen activator (u-PA) is secreted as the relatively inactive pro-urokinase; it is converted to the active form (single-chain urokinase) by plasmin. Single-chain urokinase is converted to its most active form (double-chain urokinase) by kallikrein, which is released during activation of the coagulation cascade.
Plasmin-mediated fibrinolysis is confined to the clot by the local availability of fibrin and by plasminogen activator inhibitor-1 (PAI-1), which is secreted by many cells, and by plasminogen activator inhibitor-2 (PAI-2), which is secreted primarily by the placenta. Thrombin-activatable fibrinolysis inhibitor (TAFI) is synthesized in the liver, is activated by the thrombin-thrombomodulin complex, and inhibits fibrinolysis by eliminating the binding sites on fibrin for plasminogen and t-PA. The antifibrinolytic drugs tranexamic acid and epsilon-aminocaproic acid (EACA) inhibit fibrinolysis by binding to plasminogen and plasmin and preventing their binding to fibrin.
Changes in the concentrations of coagulation factors during pregnancy are outlined in Chapter 2 (see Box 2-2). The levels of most procoagulants increase during pregnancy, while anticoagulant levels remain stable or decrease.64 Although t-PA levels decrease and antifibrinolytic proteins (i.e., PAI-1, PAI-2, TAFI) increase, plasminogen and fibrin degradation product levels increase, suggesting that fibrinolysis continues unabated during pregnancy.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


